INTRODUCTION The Goldenhar Syndrome is a multifactorial inheritance, presenting more severe manifestations when this inheritance is paternal, autosomal dominant. It can also be identified by the similar ocular-auricle-vertebral dysplasia, OAV dysplasia, facial-auricle-vertebral sequence, FAV sequence, ocular-auricle-vertebral spectrum.
This pathology has as classic triad the ocular, auricular and vertebral alterations, being able to present systematic signals, with cardiac, renal and central nervous system anomalies(1,2,3,4,5). It originates from a vascular disruption in the embryo, between 35th and 40th day of gestation, hindering correct morphogenesis of structures derived from the first and second bronchial arcs, resulting in the clinical picture which is present at birth(2).
This illness is recognized as a syndrome because, besides mandibulofacial dysostosis, the additional presence of epibulbar dermoid and vertebral anomalies exists(6). The incidence is one every 56,000 births(7), bilaterally predominating in men, being able to be associated to genetic, chromosomal and teratogenic environmental disorders. In North Ireland the prevalence tax is one every 45,000 live births(8). Familiar cases with dominant autosomal inheritance with changeable expressivity have already been reported, in addition to cases of consanguinity between parents, suggesting recessive autosomal inheritance. Hemifacial microsomia and the dysplasias of auricular pinna are present in 65% of the cases, epibulbar tumors and the congenital cardiopathies in 50% of the cases, being the Tetralogy of Fallot the most frequent one(9). Up to 30% of the patients present column alterations, varying from bifid spine and hemivertebra up to vertebral fusing and hypoplasia. In 5% renal and trachea alterations occur. It is estimated that that 5% to 10% have some degree of mental deficiency, with or without structural alteration of the central nervous system.
In this syndrome a congenital symptomatic complex of unknown etiology occurs, in which the main alterations are located in the eye (epibulbar dermoid and/or lipodermoid), in the external ear (blind auricular appendices, fistulas) and in the vertebral column (hemivertebras, vertebral fusing and other diverse malformations), being also associated to other visceral or face congenital malformations involving structures derived from 1st and 2nd branchial arcs(10). It is caused by multifactorial inheritance, having generally unilateral alterations at continuous development(1). When bilateral, one of the sides is more affected(2).
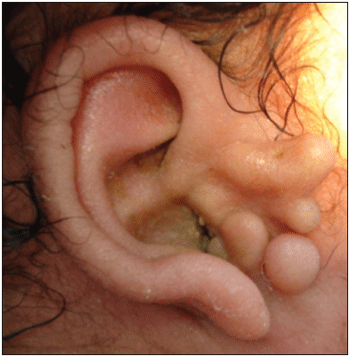
Figure 1. Goldenhar Syndrome-Right auricular pinna dysplasia, with the presence of pré-auricular apêndices.
The diagnosis of the Goldenhar Syndrome shouldn't be based only on radiological or laboratorial results. It must be based on clinical aspects and associated with systemic conditions of birth and on radiological findings(11).
The therapeutical strategy is directly related to the type of affection, uni or bilateral, thus being able to use surgical procedures or other specific methods(12).
The day-by-day interdisciplinary performance present at the Hospital Universitário Bettina Ferro de Souza motivated us to a deeper study of the congenital anomalies, amongst which the Goldenhar Syndrome, among the Otorhinolaryngology, Ophthalmology, Pediatrics, Psychology, Social Service and Physiotherapy clinics, mainly referring to assistance to children carrying several special necessities. The accomplishment of this study is justified for the necessity to alert the health professionals for the occurrence of this syndrome, which is little frequent, but has serious consequences to its carriers, mainly when exposed to a late diagnosis. This subject is widely studied in the gratuitous data base Online Mendelian Inheritance in Man - OMIM, which brings information and articles constantly brought up to date(13).
REVISION OF LITERATURE
Etiopathogeny The multifactorial etiopathogeny includes nutritional and environmental factors that result in blastogenesis disorders. Information to identify these etiological factors do not exist(14). Chromosomal abnormalities are verified and theories suggest a disorder of the superior part of neural cells as the cause of the illness. The influence of other factors, including the environment during the pregnancy can be responsible for the appearance of the illness. The ingestion of some drugs such as cocaine, thalidomide, retinoic acid and tamoxifen by mothers is reported to be factors for the development of the illness.
Although new cases with dominant autosomal inheritance have been described, it is believed that most of them is sporadic. On the other hand, the anomalies of 1st and 2nd branchial arcs have been observed in children born from mothers who were exposed to thalidomide, primidone and retinoic acid, in addition to diabetic mothers(15,16). Embryologically, the ocular-auricle-vertebral defect has been considered an anomaly of 1st branchial arc, but this alteration does not explain the anomalies in the brain, heart, kidneys or dorsal spine(17).
Important association between the development of this syndrome and the presence of malnutrition, contact with tobacco and herbicide was verified, which lead to the production of free radicals that provoke the DNA break resulting in congenital malformation.
Clinical manifestations The classical characteristic triad of this syndrome is formed by ocular, auricular and vertebral alterations(18). They can be associated to: malar and/or mandibular hypoplasia; hypoplasia of the facial musculature; micrognathia; pre-auricular appendices and dysplasia of external ear; hemivertebra or hypoplasia of cervical, thoracic or lumbar vertebrae; epibulbar dermoid; microphtalmy; palate and/or cleft lip; cardiac anomalies; renal anomalies and central nervous system anomalies. However, due to variability of clinical picture, there are patients who are affected with minimum clinical manifestations predominating face asymmetry and the dysplasia of auricular pinna.
Pre-auricular lumps or fistulas can be found as first manifestation, followed by the orthopedic alterations. The most frequent bone abnormalities are of cervical vertebral, as occipitalization of Atlas, synostosis, cuneiform vertebrae, and the affection of more inferior vertebrae has great incidence, similar to what occurs in the bifid spine. Another frequent affection is the ocular ones, as the alteration of the epibulbar dermoid.
Post-mortem description data of 1-and-2-year-old patients have also been found, where absence of the olfactory foramen in the lamina cribosa of the ethmoidal and ipsilateral bone of the olfactory bulb and tract were verified.
There are important associations with cardiac, pulmonary and renal alterations predominantly evidenced in the first months of life, but there are diagnosis reports in adult patients with more than thirty years of age. Rare affections as lipoma of callous body have been described(1).
According to genetic findings, the manifestations are generally more serious when the inheritance is paternal, autosomal dominant, evidence of genetic heterogeneity and may also appear. The patients present greater susceptibility for diabetes, hyperlipidemia and for the embryopahtological effect caused by rubella. Some structural alterations of the temporal bone individually facilitate the occurrence of meningitis and posterior affection of ears.
The auricular malformations can be unilateral or bilateral, appearing pinna umbilication, which can be implanted previously or inferiorly, besides its form varying from micrognathia up to the lobe widening or even anognatia. In some cases they can coexist with periauricular fistulas. The external auditory canal can be presented with stenosis, agenesia or have the auditory canal finishing in cul-de-sac. Mastoiditis is also observed.
When the exploration of the oral cavity was made, the characteristics of the palate are verified, whose alterations show relation to possible disorders of auditory tube and the ventilation of the medium ear.
The characteristics of hair, eyebrow, eyes, nose and neck implantation are observed, revealing association with dysgenetical syndromes(19).
Many patients have been reported presenting multisystemic complex anomalies, associated to Goldenhar Syndrome, including respiratory, dermatological, bone, genital, urinary, gastrointestinal, muscular and central nervous systems and endocrinal system problems(20). The frequency of the cardiovascular malformations in the Goldenhar syndrome varies from 5% to 58%. The main reason for this variation is the adoption of different diagnostic criteria. Congenital cardiac problems and tetralogy of Fallot are the most frequently found cardiac malformations in this syndrome. Other reported cardiovascular problems include coarctation of aorta, malformation of arterial ducts, transport inside of important arteries, dextrocardia, atrial, septal and ventricular defects, anomalies of coronary arteries, abbaissement of aorta to the right side, anomalies of the upper/lower cava veins and arrhythmic disorders similar to what happens in the syndrome of Wolff-Parkinson-White(21).
Complementary diagnosis and Examinations Currently, this diagnosis can be established since the prenatal phase. A recent study detected multiple congenital anomalies in ecography findings in 20 cases. The gestational age in the diagnosis varied from 15 to 35 weeks and in more than half of the cases it was associated to the polyhydramnios or oligohydramnios. The characteristics observed were: face malformations present in 52% of the cases, as microphtalmy, auricular abnormalities, face asymmetry and face cleft; amongst the neurological alterations, occiptal encephalocele, hydrocephaly and cerebellar hypoplasia were observed, which occured in 47% of the findings; cardiac defects, radial aplasia and renal agenesia were seen less frequently(9,22).
In terms of diagnosis after birth, there is a global consensus that the diagnosis of the Goldenhar Syndrome does not have only to be based on radiological or laboratorial results, the clinical aspects and the ones associated with systemic conditions of birth and radiological findings must also be considered(11). Most of the authors consider the presence of ear anomalies (microtia) and appendices in the ear necessary for diagnosis. Moreover, facial or mandibular hypoplasia facial asymmetry, dermoid epibulbar tumor, palpebral alterations, vertebral anomalies, lateral face fissuring and renal problems are also observed(23,24).
Both laboratorial and image tests are important for the diagnosis, once they are directly related to provoked alterations. The X-ray of the zigomatic bones shows a macroscopic deficiency of bone symmetry development. Also a possibility of agenesia of these bones exists, with defect of fusing of the zigomatic arc and agenesia of the palatine bones. Palatine cleft must radiographically be observed. Ophthalmological and otorhinolaryngological examinations are also important for the final diagnosis(25).
The audiological evaluation comprehends the Pure-Tone Audiometry, Immitanciometry, Brainstem Evoked Response Audiometry (BERA) and Transient Evoked Otoacoustic Emissions and Distortion Product Otoacoustic Emissions.
It is necessary to carry through the functional examination of audition for a precocious detention of the malformations, moth unilateral and bilateral. It is observed that mainly conductive hypoacusis between 50dB and 70dB, being able to coexist with malformation of the internal ear, translated into mixing hypoacusis, with auditory losses between 80dB and 90dB.
The radiological examination of choice is the CT scan of the mastoid, which informs us on the state of the medium and internal ears and on the state of mastoid pneumatization. These examinations must be carried through after three years of age. The accomplishment of genetic studies is important, as well as to discard malformations in others systems(26).
Distinguishing diagnosis Due to the clinical heterogeneity, it is important to focus other diagnostic possibilities, as the associations with the Townes-Brocks and branchial-oto-renal syndromes. The distinguishing diagnosis with these other entities is made based on the standard of the anomalies found(27).
When there are cardiovascular malformations and/or symptomatology that indicates associated cardiac problem to other classic characteristics of the syndrome of Goldenhar or not, it is necessary to make distinguishing diagnosis with other genetic problems which present similar findings as Willians Syndrome, Ehlers-Danlos Syndrome and Fabry Disease(28).
Treatment In terms of the auricular alterations, the use of the correct therapeutical strategy must consider if the malformation is unilateral or bilateral and if it presents association with another dygenetical syndrome.
In case the alteration is unilateral it is recommended to do the surgery at five years of age, which should be carried through at two moments, the first one with the aesthetic reconstruction, by the plastic surgeon or by the otorhinolaryngologist himself, and secondly the functional reconstruction, according to the development and aeration of the medium ear and mastoidal pneumatization. This is due to alterations in the distensibility of the skin for cicatricial tissue formation and difficulties of mobilization of the skin when the aesthetic surgery is made after the functionary one. In the patients with bilateral syndrome the precocious auditory stimulation must be carried through the bone way. To evaluate the development of the language, beyond evaluation and control of the superior air ways. If this is the case, surgery is indicated from three years of age on, and the main surgical techniques used are: Patee Technique, Wullstein-Zoellner Technique, Ombredanne Technique, Tato Technique and Diamond Technique(12).
Another concern is with the permeability of the respiratory tract. The aforementioned malformations sometimes imposes the necessity of tracheostomies or, craniomaxilofaciaal maneuvers.
CONCLUSION The rarity of Goldenhar Syndrome and its heterogeneity of spectrums demonstrate the multifactorial feature of this pathology. Having as classic triad the ocular, auricular and vertebraal alterations, they present the hemifacial microsomia and the dysplasias of auricular pinna as more frequent alterations. Once it is probably a multifactorial dysgenetical syndrome, the genetic counseling and the detailed study of the cause in each patient affected by this syndrome is necessary, once associated factors such as ingestion of drugs during the gestation, gestacional diabetes, alcohol ingestion during the pregnancy, amongst others, can be prevented, avoiding the appearance of new cases as much as possible.
BIBLIOGRAPHICAL REFERENCES1. Kalavrezos ND, Bochlogyros PN, Panos G. - Surgical corrections of a pacient with Goldenhar syndrome. Heel Period Stomt Gnathopathoprosopike Cheir, 1990; 5(4):147-50.
2. Gorlin RL. Brachial arch and oro-acral disorders. In: Gorlin JJ, Cohen Jr MM, Hennekam RC, editors. Syndromes of the head and neck. London: Oxford University Press; 2001. p.790-7.
3. Bestlmeyer U, Weeda H, Siegert R, Greiwe Schwinger E. Familial Ocurrence of Oculoauriculovertebral Dysplasia and Franceschetti Syndrome. Hno,1996; 44(8):252-5.
4. Altamar RJ. Sindrome de Goldenhar - a propósito de um caso. An. Otorrinolaringol. Iber Am. 1998; XXV:491-7.
5. Regenbogen L, Godel V, Goya V, Goodman RM. Further evidence for an autosomal dominant form of oculoauriculovertebral dysplasia. Clin.Gen 1982; 21:161-7.
6. Hunt JA, Hobar PC. Common craniofacial anomalles: the facial dysostoses. Plat Reconstr Surg. 2002; 110:1714-25.
7. Gorlin JJ, Cohen MMJr, Levin LS. Syndromes of the Head and Neck. 3rd ed. London: Oxford Univ. Press 1990. Pp. 641-649.
8. Morrison PJ, Mulholland HC, Craig BG, Nevin NC. Cardiovascular abnormalities in the oculo-auriculo-vertebral spectrum (Goldenhar syndrome). Am J Med Genet. 1992 Nov 1;44(4):425-8.
9. Verona LL,Damian NG, Pavarina LP, Ferreira CHF, Melo DG. Síndrome de Goldenhar: relato de um caso com discordância em gêmeas monozigóticas. J Pediatria Vol. 82, Nº1, 2006.
10. Laredo FJ, Braga JMB, Kasinsk SK, Caballero JMP. Síndrome de Goldenhar (displasia óculo-aurículo-vertebral). Folha Med. Bras. 1985; 91:361-4.
11. Araneta MRG, Moore CI, Onley RS, Edmonds LD, Karcher JA, McDonough C, Hiliopoulos KM, Schlangen KM, Gray GC. Goldenhar syndrome among infants born in military hospital to Gulfwar veterans. Teratology1997; 56:244-251.
12. Alonso JM, Tato JM. Tratado de Otorrinolaringologia e broncoesofagica. 4º edição.
13. OMIM. Online Mendelian Inheritance in Man. Hemifacial Microsomia [On line]. Disponível em
http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=164210. Acessado em 22 de abril de 2007.
14. Lessick M, Vasa R, Israel J. Severe manifestations of oculoauriculovertebral spectrum in a cocaine-exposed infant. J Med Gen1991; 28:803-4.
15. Gorlin RJ, Cohen MM, Levin LS. Branchial arch and oroacral disorders. In: Syndromes of the head and neck. 3ed. New York: Oxford University Press; p.641-8.
16. Johnson K, Fairhust J, Clarke NM. Oculoauriculovertebral spectrum: new manifestations. Pediatr. Radiol. 1994; 25:146-8
17. Regenbogen L, Godel V, Goya V, Godman RM. Further evidence for an autosomal dominant form of oculoauriculovertebral dysplasia. Clin Genet. 1982; 21:161- 7.
18. Brosco KC, Zorzetto NL, Costa AR. Perfil audiologico de indivíduos portadores da síndrome de Goldenhar. Rev. Brás. Otorrinolaringol. 2004; 70(5):645-9.
19. Cotin G, Lacombe H. Malformations du Condut-auditif externe et de láreile moyenne. EMC. 20182-A-10. 1985.
20. Rollnick BR, Kaye CL, Nagatoshi K, Hauck W, Martin AO. Oculoauriculovertebral dysplasia and variants: Phenotypic characteristics of 294 patients. Am. J. Med. Genet. 1993; 26:361-75.
21. Greenword RD et al. Cardiovascular malformations in oculoauriculovertebral dysplasia (Goldenhar syndrome). J. pediatr. 1974; 85:816-8.
22. Castori M, Brancati F, Rinaldi R, Adami L, Mingarelli R, Grammatico P, Dallapiccola B. Antenatal presentation of the oculo-auriculo-vertebral spectrum (OAVS). Am. J. Med. Genet. 140A: 1573-1579, 2006.
23. Ostlere SJ, MacDonald B, Athanasou NA. Mesenchymal chondrsarcoma associated with Goldenhar´s syndrome. Arch. Orthop. Traum. Surg. 1999; 30:120-4.
24. Llano-Rivas I, Gonzalez AA, Castillo V, Reyes R, Carnevale A. Microtia: a clinical and genetic study at the National Institute of Pediatrics in México city. Arch. Méd. Res. 1999; 30:120-4.
25. Schaffer AJ, Hine MK, Levy BM. Tratado de patologia bucal. ed.4.Rio de Janeiro: Interamericana; 1985. p.631.
26. Tramba H, Herman P. Formes cliniques dês OMC non cholesteatoma. Encyclopedie Medico-Chirugicale (Paris- France) 20.095-A-10.1993.
27. Rollnick BR. Oculoauriculovertebral anomaly: variability and causal heterogeneity. Am. J. Méd. Genet. Suppl. 1988; 4:41-53.
28. Francois B, de Paepe A, Matton MT, Clement D. Pulse wave velocity recordings in a family with ecchymotic Ehlers- Danlos syndrome. Int. Angiol. 1986; 5(1):1-5.
1. Master's Degree in Otorhinolaryngology by Universidade Federal do Rio de Janeiro and Doctor's degree student on Neuroscience by Universidade Federal - Pará (Aid Professor at Universidade Federal - Pará and at Universidade do Estado do Pará)
2. Otorhinolaryngology Monitor at Universidade do Estado do Pará (Junior undergraduate student in Medicine at Universidade do Estado do Pará)
3. Sophomore undergraduate student in Medicine at Universidade do Estado do Pará.
Universidade do Estado do Pará - UEPA
Francisco Xavier Palheta Neto
Adress: Travessa Barão do Triunfo, 3380 Apt 502 - ZIP: 66093-050 - Belém/PA - Phone: (91) 3224-6517 / (91) 9116-0508 - Phone/Fax: (91) 3236-0227 - E-mail: franciscopalheta@hotmail.com
This article was submitted to SGP (Sistema de Gestão de Publicações) of R@IO on January 23, 2007 and approved on May 26, 2007 at 07:48:16.

