INTRODUCTIONThe classic account, made by DANDY & BLACKFAN in 1914*, where autopsies showed severe hypertentorial hydrocephalus, cystic dilatation of the fourth ventricle, small vermis, removal of the cerebelar hemispheres and absence of the roof of the fourth ventricle, thickening and opacity of the pia-arachnoid cisternae of the skull base and dilatation of the aqueduct (1).
The DANDY-WALKER syndrome (SDW) is a non-familial syndrome characterized by cystic dilatation of the fourth ventricle and aplasia or partial or total atrophy of the cerebelar vermis. Usually presents atresia of foramina of MAGENDIE and LUSHKA. In 75% of cases occur other brain malformations such as agenesis of the corpus callosum, heteropsias, lissencephaly, stenosis of the aqueduct of SYLVIUS (2-3). GARDNER et al** proposed that the SDW, along with other syndromes (Arnold-Chiari malformation, cerebelar arachnid cyst and syringomyelia) were manifestations of the same disease (1).
Some studies show an incidence of approximately 70% of relationship between the SDW and systemic abnormalities (1). Little is known about congenital malformations of the posterior pit structures, their genetic alterations have been mapped to chromosome 3q (4-5), but the gene is not located exactly, but it is known that the basis of the development process of the structures of the pit later is nature for human cerebelar malformations (5). It is also known that structures cerebelar growth early embryo in the period until the early post-natal, this event would cerebellum susceptible to a wide spectrum of disorders of development (6).
The pathogenesis of this syndrome is controversial, but the most accepted theory is that the leaf development of the foramina of MAGENDIE and LUSHKA during the fourth month of fetal life, leading to bulging cystic fourth ventricle. New theories have proposed that the SDW result from a failure to develop the roof of the hindbrain, taking this as a cause teratogenic effect (1). Some studies suggest that the use of warfarin in the long run would be responsible for the development of SDW in 1-2% of exposed fetuses (7). What further strengthens this theory is that a study by HART et al., Showed no relationship between degree of hydrocephalus and the size of the cyst in the posterior pit, vermis or attenuation of the permeability of the fourth ventricle and hydrocephalus in some cases was absent (1).
The SDW is an entity of heterogenic hyperplasia of the cerebelar vermis and is a newly identified gene associated with the connection with X-HPRT (8) also relates to the basal ganglia disease.
Clinically there may be moderate delayed psychomotor development, microcephaly, hypotonia, but the predominant symptoms refers to hydrocephalus, usually in the first two years of life, this however can be ignored, appearing late (first or second decade of life) (2 - 9). Hydrocephalus occurs by obstruction of the foramina and LUSHKA MANGEDIE (1). Some ocular abnormalities are described in the SDW, as coloboma corioretinian (10), nystagmus (11-13). There may be mental retardation (50%), spasticity (instead of hypotonia), seizures, vomiting, all depending on the degree of cerebelar malformation (3). In patients with vermis fissures with two conformations and almost normal brain functions are also almost normal without association with other malformations. In patients with severe malformations of the cerebellum, vermis with only one or no crack, it is common to severe mental retardation and other malformations of the central nervous system, such as agenesis of the corpus callosum. It is divided, so the SDW into two large groups according to the previous malformations for determining the prognosis intellectual (12-13). It is reported in the literature of the coexistence of large facial coetaneous hemangioma with SDW (14). Other syndromes where coexist brain and ocular malformations are reported as Neuhauser syndrome (MMMM - megalocornea, macrocephaly, mental and motor retardation), which are found cortical atrophy, enlargement of the fourth ventricle, hyperplasia of the corpus callosum. All this considered a variant of SDW with megalocornea (15). For diagnosis, there is need for magnetic resonance images of good quality axial view of the cerebelar vermis and T2 images (12). The neuroradiological findings are characteristic, such as cystic dilatation of the fourth ventricle and changes in the cerebelar vermis (2), besides others already mentioned. The SDW should always be accompanied by a multidisciplinary team. The treatment of hydrocephalus is always surgical through a ventricle-peritoneal shunt (2).
The bilateral sensorineural deafness can be part of the clinical picture of the syndrome and we describe a case whose patient underwent implantation cochlear by presenting profound bilateral deafness.
The aim of this paper is to describe a case of female patient, 13 years with a diagnosis of this syndrome and bilateral hearing loss underwent cochlear implant surgery under local anesthesia and sedation.
CASE REPORTCGS, 13 years old female was referred to the ENT Department of Otolaryngology Institute of Parana with a diagnosis of "Dandy-Walker syndrome" for ENT evaluation for bilateral hearing loss with no response to the use of hearing aids.
The patient performs follow up with neurology since birth, with a history of gestational rubella, cataracts in both eye and neurological surgery prior hydrocephalus, absence of parental consanguinity. Child with imaging exam compatible with Dandy-Walker syndrome with posterior pit collection and communication with the fourth ventricle, associated with signs of hyperplasia of the cerebelar hemispheres and vermis. Karyotype not performed. Deficit neuropsychomotor light.
The child met criteria for cochlear implant. She underwent by otolaryngology, neurology, speech and psychological medical evaluation.
Audiometry showed profound sensory neural hearing loss bilaterally. In electrophysiological assessment of hearing was not possible to detect brainstem evoked potentials bilaterally. In the absence of otoacoustic emission response bilaterally MRI of the brain showing cerebelar vermis hyperplasia associated with mega cistern magna, communicating with the fourth ventricle (Figures 1 and 2).
The resonance settings ears inner ears are symmetrical and normal configuration. There are no anomalous enhancements related to them or the rest of the brain. The fluid spaces of the cochlea are normal thickness, ranging from 1.3 to 1.7 mm the right and 1.4 to 1.7 mm left. The cochlear nerves have been identified and normal thickness of 0.6 mm (Figure 3).
She was underwent a cochlear implant in her right ear. The prosthesis used was a nucleus freedom implant of the Cochlear Comparation®. Surgery was performed under local anesthesia and sedation by retroauricular access, creation of niche outdoor unit, closed mastoidectomy, posterior tympanotomy, chocleostomy, inserting the internal components and telemetry. All steps performed uneventfully. The implant was activated 1 month after surgery with excellent sound perception at the time of activation. The patient has excellent levels of discrimination with less need for lip reading and excellent response to therapy with spectacular mend the quality of speech.
No doubt the results bring impact on quality of life of patients while providing better social integration.
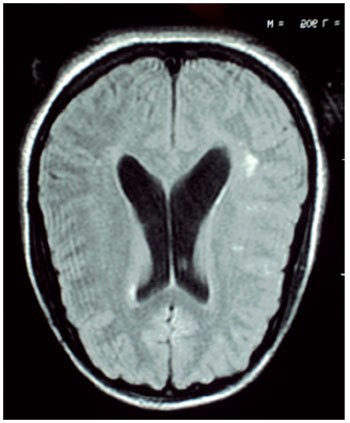
Figure 1.
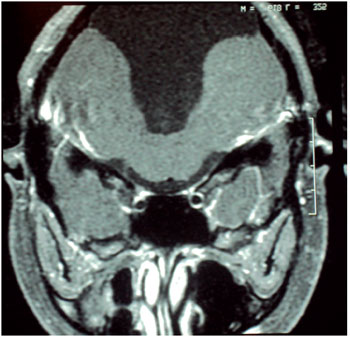
Figure 2.
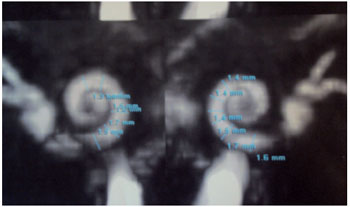
Figure 3.
The field of cochlear implants is growing rapidly due to improved quality of the implants, surgery less invasive, and wider dissemination of this type of treatment of deafness. Cochlear implants are extremely expensive prostheses that partially replace the functions of the cochlea (20). The surgery is now much faster and less invasive than some years ago, with smaller incisions and less morbidity for the patient.
The patient even though the syndrome Dandy-Walker does not present significant deficit in neuropsychomotor development, fulfilling the criteria established in the literature for the performance of cochlear implant surgery. It Was evaluated by a multidisciplinary team with no contraindications for the procedure. There are already cases of patients with the same syndrome who underwent cochlear implant surgery, complications described in the literature (21) and with good results despite co morbidities.
In our facility we chose to perform this surgery under local anesthesia and sedation. This type of surgical anesthesia brings morbidity / anesthetic besides a minor post-operative recovery and faster lower hospital costs when compared with general anesthesia and were perfect forward from the patient.
We believe that the presence of Dandy-Walker syndrome cannot be considered a contraindication to the performance of cochlear implant surgery, and there were no surgical complications due to neurological disorders with very favorable results for the patient who exhibits excellent discrimination. It has less need for lip reading with improvement in speech quality.
REFERENCES1. Diament A. Neurologia infantil 3a ed. São Paulo: Atheneu; 1996.
2. Rosenberg S. Neuropediatria. São Paulo: Sarvier; 1995.
3. Yilmaz MA. The site on Dandy Walker Syndrome [homepage on the Internet]. [cited 2005 Aug 29]. Available from: http://www.geocities.com/murat_yil/dandy.html
4. Parisi MA, Dobyns WB. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab, 2003; 80(1-2):36-53.
5. Chizhikov V, Millen KJ. Development and malformations of the cerebellum in mice. Mol Genet Metab. 2003, 80(1-2):54-65.
6. Ten Donkelaar HJ, Lammens M, Wesseling P, Thijssen HO, Renier WO. Development and developmental disorders of the human cerebellum. J Neurol. 2003, 250(9):1025-36.
7. Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med. 1998, 338(16): 1128-37.
8. Daufenbach DR, Ruttum MS, Pulido JS, Keech RV. Chorioretinal colobomas in a pediatric population. Ophthalmology. 1998, 105(8):1455-8. Comment in: Ophthalmology. 1999, 106(4):645-6.
9. Niesen CE. Malformations of the posterior fossa: current perspectives. Semin Pediatr Neurol. 2002, 9(4):320-34.
10. Coats DK, Paysse EA, Levy ML. PHACE: a neurocutaneous syndrome with important ophthalmologic implications: case report and literature review. Ophthalmology. 1999, 106(9):1739-41.
11. Kumandas S, Akcakus M, Coskun A, Gumus H. Joubert syndrome: review and report of seven new cases. Eur J Neurol. 2004, 11(8):505-10.
12. Klein O, Pierre-Kahn A, Boddaert N, Parisot D, Brunelle F. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst. 2003, 19(78):484-9.
13. Boddaert N, Klein O, Ferguson N, Sonigo P, Parisot D, Hertz-Pannier L, et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology. 2003, 45(5):320-4.
14. Reese V, , Paller AS, Esterly NB, Ferriero D, Levy ML, et al. Association of facial hemangiomas with Dandy-Walker and other posterior fossa malformations. J Pediatr 1993, 122(3):379-84.
15. Balci S, Teksam O, Gedik S. Megalocornea, macrocephaly, mental and motor retardation: MMMM syndrome (Neuhauser syndrome) in two sisters with hypoplastic corpus callosum. Turk J Pediatr. 2002, 44(3):274-7.
16. Pagon RA, Clarren SK, Milam DF Jr, Hendrickson AE. Autosomal recessive eye and brain anomalies: Warburg syndrome. J Pediatr. 1983, 102(4):542-6.
17. Golden JA. Cell migration and cerebral cortical development. Neuropathol Appl Neurobiol. 2001, 27(1):22-8.
18. Ruibal Francisco JL, Sánchez Buron P, Piñero Martinez E, Bueno Lozano G. [Turner's syndrome. Relationship between the karyotypes and malformations and associated diseases in 23 patients]. An Esp Pediatr. 1997, 47(2):167-71. Spanish.
19. Kawaguchi T, Jokura H, Kusaka Y, Shirane R, Yoshimoto T. Intraoperative direct neuroendoscopic observation of the aqueduct in Dandy-Walker malformation. Acta Neurochir (Wien). 2003, 145(1):63-7
20. Chakrabarty A, Tarneja VK, Singh VK. Cochlear implant: anesthesia challenges. MJAFI Hournal, 1994, 60:351-6.
21. Sharon LC*, Laurie Mb, Evan J, Alok S, Tracy S. Successful cochlear implantation in a child with Keratosis, Icthiosis and Deafness (KID) Syndromeand Dandy-Walker malformation. International Journal of Pediatric Otorhinolaryngology. 2008;72,693-698.
1) Otolaryngological Doctor at Hospital of Parana of Otolaryngology.
2) Professor of Otolaryngology, Federal University of Parana. Otolaryngologist Doctor at, Hospital of Parana of Otolaryngology.
3) Head of the Department of Otolaryngology, Clinics Hospital, Federal University of Parana. Head of the Department of Otolaryngology, Clinics Hospital, Federal University of Parana.
4) Resident of Otolaryngology at the Clinics Hospital, Federal University of Parana.
Institution: University of Parana in otolaryngology. Curitiba / PR - Brazil. Mailing address: Adriana de Oliveira Kosma Pires - Rua Edmundo de Amicis, 316 - Curitiba / PR - Brazil - CEP: 81810-160 - E-mail: adri_pires@hotmail.com
Manuscript received June 2, 2010. Article accepted on August 1, 2010.

