INTRODUCTIONBullous systemic lupus erythematosus (BSLE) is an autoantibody-mediated disease with subepidermal blisters. It is a rare presentation of SLE occurring in less than 5% of lupus cases (1). A study conducted in 3 regions of France reported an incidence of 0.2 cases per million inhabitants (2). Although the disease affects both men and women regardless of race or age, it occurs more frequently in black women between the second and third decades of life.
Clinically, BSLE presents as tense bullous lesions that can be serous or hemorrhagic and spread rapidly to all parts of the body, although lesions are mainly found on the trunk and in sun-exposed areas; they can also affect mucous membranes. These lesions often indicate lupus manifestation (1). However, they must be differentiated from other bullous dermatoses such as epidermolysis bullosa acquisita (EBA), dermatitis herpetiformis, bullous pemphigoid, and linear IgA bullous dermatosis (3).
In this paper, we report the case of a young female patient who suddenly developed bullous disease and was admitted to hospital because of severe oral lesions. After immunofluorescent examination, a diagnosis of bullous lupus erythematosus was made.
CASE REPORTHere, we report a 27-year-old, single, catholic, female, born in Sierra Taboão and raised in Embu. The patient reported emergence of painful bullous lesions on the nasal ala and left buccal mucosa that displayed sudden and rapid growth (Fig. 1). She sought advice from the emergency dermatology staff 15 days after the onset of symptoms and was hospitalized with suspected bullous disease. General examinations were normal and her leukocyte levels were unchanged. Upon hospitalization, intravenous steroids were introduced but the patient showed no clinical improvement. She displayed further extensive injuries to the trunk, axillae, and vulva as well as rupture of the bullous lesions, which remained as reddened scars.
We performed an incisional biopsy of a lesion in the left buccal mucosa. The pathology results indicated mucositis with extensive erosion and the presence of a predominantly neutrophilic inflammatory infiltrate with degeneration and apoptosis of basal layer keratinocytes (Fig. 2). Direct skin immunofluorescence showed anti-IgA, anti-IgM, and anti-IgG linear fluorescence on the continuous dermal side of the cleavage (Fig. 3).
Indirect immunofluorescence of the skin showed conjugated anti-IgA, was anti-IgM negative, and indicated pemphigus. It also displayed anti-IgG conjugated with fluorescence in the nucleus of keratinocytes, confirming the diagnosis of bullous lupus erythematosus.
As initial treatment, we administered 100 mg dapsone, 250 mg chloroquine, 50 mg azathioprine, and 50 mg prednisone. Although this resulted in improvement of the lesions, there was persistence of some scarring plaques on the trunk and lower lip. The patient was followed up for 6 months. During this period, she remained clinically stable, without active lesions in the oral mucosa and lips (Fig. 4), and we gradually reduced her dose of prednisone.
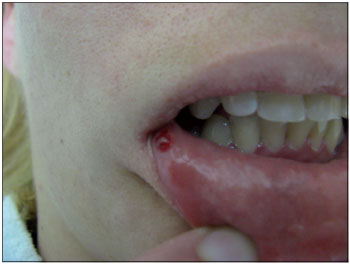
Figure 1. Bullous lesion in the right lower lip.
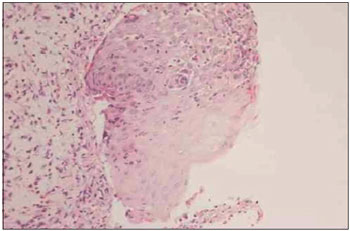
Figure 2. Predominantly neutrophilic inflammatory infiltrate with hydropic degeneration of the basal layer and apoptotic keratinocytes.
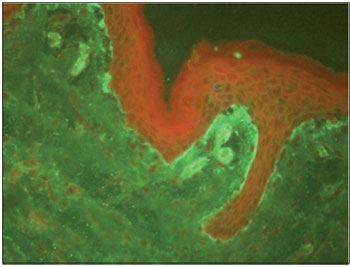
Figure 3. Direct immunofluorescence of the skin showing linear fluorescence on the continuous dermal side of the cleavage.
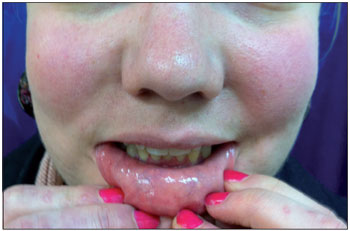
Figure 4. Lack of oral lesions, without mucosal scarring.
In 1973, Pedro and Dahl (4) described the first case of BSLE. After this, several cases with similar characteristics were reported. BSLE patients produce autoantibodies that recognize type VII collagen, a major component of anchoring fibrils, which play an important role in dermoepidermal adhesion.
Chan and colleagues (5) identified further autoantibodies reacting to multiple basement membrane components including bullous pemphigoid antigen 1, laminin-5, and laminin-6 in patients with BSLE. This hyperimmune state seems to be associated with the gene for major histocompatibility complex HLA-DR2 (4).
The clinical manifestations of BSLE are characterized by vesicles, serous blisters, or hemorrhagic content in the face, neck, and trunk. Lesions can be located in areas both exposed and unexposed to the sun. They may be accompanied by mild to severe itching and can affect mucous membranes. BSLE lesions may heal completely, or result in hypo-or hyperpigmentation or scarring (6).
The clinical picture of BSLE is similar to bullous pemphigus, being distinguished by indirect immunofluorescence. Histologically, the condition presents as subepidermal blisters accompanied by neutrophil microabscesses in the papillary dermis and dermal edema. There are also large deposits of mucin and the absence of eosinophils, which helps to differentiate between dermatitis herpetiformis Duhring, linear IgA dermatosis, and EBA (6).
In BSLE, direct immunofluorescence shows deposits of immune complexes (IgG, IgA, IgM, and complement) along the basement membrane that can be granular to linear (7).
Using indirect immunofluorescence, and according to the autoantibodies present, BSLE can be classified into 3 subtypes: type 1, which reacts against collagen VII; type 2, where the location of the antigen is undefined or the dermal antigen is one other than type VII collagen; and type 3, where the antigen is epidermal (6).
BSLE responds well to treatment with dapsone, which differs from EBA. In some cases where high doses of corticosteroids alone were used for control of visceral manifestations, there was no improvement of skin lesions until the introduction of dapsone. It is expected that within 24-48 h of the introduction of dapsone, the emergence of new bubbles will be stopped, and total regression should occur within weeks. In cases that do not respond to dapsone, the use of prednisone and azathioprine is suggested. BSLE may regress completely and independently of systemic involvement without recurrence (8).
REFERENCE1. M Tincopa, Puttgen KB, Sule S, Cohen BA, Gerstenblith MR. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010 Jul-Aug;27(4):373-6.
2. Bernard P, Vaillant L, Labeille B, et al. Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol. 1995;131(1):48-52.
3. Vieira FMJ, Oliveira ZNP. Bullous systemic lupus erythematosus. An Bras Dermatol. 1998;73:143-7.
4. Peter SD, Dahl MV. Direct immunofluorescence of Bullous systemic lupus erythematosus. Arch Dermatol. 1973;107:118-20.
5. Chan LS, LaPiere JC, Chen M, Traczyk T, Mancini AJ, Paller AS, et al. Bullous systemic lupus erythematosus with autoantibodies recognizing multiple skin basement membrane components, bullous pemphigoid antigen 1, laminin-5, laminin-6, and type VII collagen. Arch Dermatol. 1999;135:569-73.
6. Obermoser G, Sontheimer RD, Zelger B. Overview of common, rare and atypical manifestations of cutaneous lupus erythematosus and histopathological correlates. Lupus. 2010 Aug;19(9):1050-70.
7. Cato, Ellen Erie et al. Bullous systemic lupus erythematosus associated with lupus nephritis: report of two cases. An Bras Dermatol. 2007;82(1):57-61.
8. Vassileva S. Bullous systemic lupus erythematosus. Clin Dermatol. 2004 Mar-Apr;22(2):129-38.
1) Associate Professor in the Department of Otorhinolaryngology, School of Medicine, University of São Paulo, São Paulo, Brazil.
2) Otolaryngologist in the Department of Otorhinolaryngology, School of Medicine, University of São Paulo, São Paulo, Brazil.
3) Specialist in Otolaryngology, School of Medicine, University of São Paulo, São Paulo, Brazil.
4) Resident in Otolaryngology, School of Medicine, University of São Paulo, São Paulo, Brazil.
Institution: Study from the Division of Clinical Otorhinolaryngology, Clinic of Stomatology, Hospital of the Faculty of Medicine, University of São Paulo.
São Paulo / SP - Brazil. Mailing address: ENT Hospital - Azis Arruda Chagury - Avenue Dr. Enéas de Carvalho Aguiar, 155 - São Paulo / SP - Brazil - Zip code: 05403-000 - Telephone: (+55 11) 3069-6226 - E-mail: azischagury@gmail.com
Article received on September 17th, 2011. Article accepted on February 5th, 2012.

