INTRODUCTIONHearing neuropathy is due to an alteration on hearing stimulus transport by any lesions of internal ciliated cells of the hearing nerve or synapsis in them (1).
These patients clinically present pre or post-lingual hearing loss, from the most different levels up to complete deafness, and they are normally distinguished as incompatible hearing with tonal threshold.
Otoacoustic emissions and cochlear microphonism are present, making normal function of external ciliated cells evident, while brainstem audiometry shows alterations from wave I present even with tonal threshold changed (1). This neural implication is also showed by absence of estapedial reflex and medial olivocochlear reflexes.
From etiological viewpoint, there are strong evidences of a genetic and/or inflamamtory cause due to its association to sensorial and motor peripheral neuropathies, such as Charcot-Marie-Tooth syndrome, Friedreich ataxia, Guillan Barrè neuropathy. What occurs on hearing system, on these cases can precede, in many years, the appearing of peripheral neuropathy. Hyperbilirubinemia, premature conditon, anoxia are also considered risky situations to hearing neuropathy development, especially to the cases of prelingual (2,3). Genetic mutations which cause non-syndromic deafness of chromosome X or mitochondrial heredity (5) had also been presented as etiological factor of hearing neuropathy.
Relations between hearing loss of neural type and mutations of OTOF gene were established by the first time in 2003 by Varga and col (6), who identify four different mutations on such gene as responsible for hearing neuropathy of recessive heredity in four Spaniard families.
OTOF gene is responsible for codification of otoferlin (a type protein), which operates on membrane transport, and though it has important role on synaptic vesicle fusion of internal ciliated cells (6,8). It was first identified in 1996, on chromosome area 2p22-23 on members of a Lebanese consanguine family with profound hearing loss of recessive heredity, what was not described as neural, and despite the fact of being listed as one of the 16 genes involved on prelingual non-syndromic hearing loss (Hereditary Hearing Loss home page, www.uia.ac.be/dnalab/hhh), its specific relation with hearing neuropathy conditions had not been previously determined (7).
In 2004, AUNA1 gene was also identified. This one is responsible for post-lingual Hearing Neuropathy conditions, of dominante autossomic heredity, on chromosome area 13q14-21 in an American family (9).
The target of this study is to describe our first case of Hearing Neuropathy caused by mutation on OTOF gene and discuss clinical implications of this diagnosis.
CASE REPORTA girl family went to DERDIC/PUCSP in 2001, reporting that the girl did not speak. At that time, she was 2 years old and had brainstem audiometry done from other institution which pointed presence of bilateral profound sensorineural deafness and also indicated device adaptation of sonorous amplification. The family denied pre, peri and neonatal antecedents and believed the child reacted to strong intensity sounds.
The girl presented herself as shy during the examination and did not show any reaction to sounds. The general and otorhinolaryngology physical exams were normal. In our institution, the protocol of evaluation is composed by audiometric exam, by brainstem audiometry and by otoacoustic emissions, and appoitment with neuropediatrician and genetic evaluation.
A new audiometry showed bilateral profound sensorineural hearing loss, with absence of estapedial reflex and tympanometric curve type A in both ears. Brainstem audiometry was done with dense and less dense clicks and, with frequency of stimuli of 3, 19.3 and 50 stimuli per second. No wave was identified on higher intensity and there was no response alteration in relation to changing on stimulus frequency, therefore, with alteration of clicks, equal images occured on the record, typical feature of cochlear microphinism, up to 4 msec to left and up to 2 msec to right (1). Otoacoustic emissions by distortion product (2) and transient (3) were bilateral and normal, what confirms hearing neuropathy diagnosis. Neuropediatric evaluation was normal.
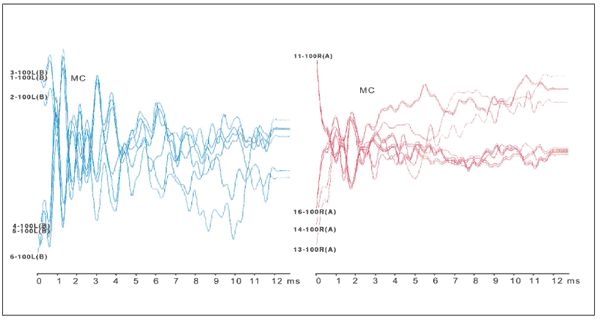 Picture 1. Cochlear microphonism present up to 4 msec at left and up to 2 msec right at
Picture 1. Cochlear microphonism present up to 4 msec at left and up to 2 msec right at 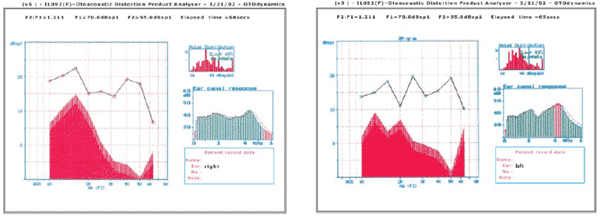 Picture 2
Picture 2. Otoacoustic emission by distortion product present at left and right.
The routine genetic study had its start with mutation tests 35delG and 167delT on Connexin 26 gene (GJB2), by the research of deletion- D13S1830 on Connexin 30 gene (GJB6), and mutation test A1555G on gene of ribosomal RNA 12S on mitochondria DNA. All these tests presented normal results. Because of hearing neuropathy condition, DNA of the patient was submitted to SSCP technique (Single Strand Conformation Polymorphism), with the purpose of screening mutations on diverse exons of OTOF gene. The migration standard of exon 15 was altered and the sequency of the same exon showed the patient has a deletion of 16 base pairs, among nucleotides 1552-1567 (1552-1567del16).
The girl is under phonotherapy focusing oral language. During therapeutical process, sonorous amplification devices with digital technology were adapted. In the beginning, it was done tests with monoaural amplifications with variations of tested ears (10), however the girl presented better performance with binaural adaptation. In relation to functinal use of hearing, she recognizes everyday sounds and can detect speech only in quiet situations; she more attentive to orofacial reading and vocalizes having few entonation variations, though with communicative intentions. She also attends special school for children with hearing impairment and has been easily learning body language.
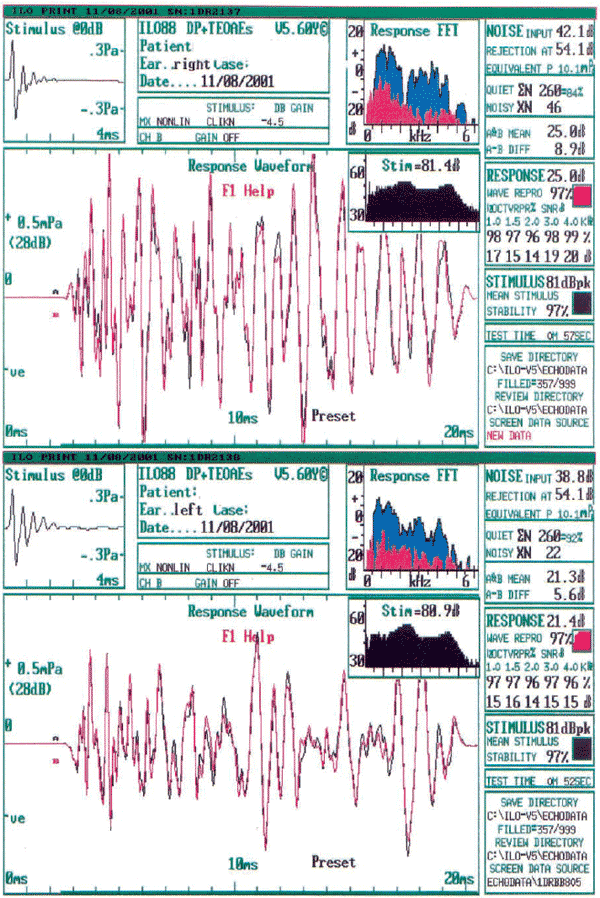 Picture 3.
Picture 3. Transient otoacoustic emission present at left and right.
A precocious or prelingual hearing neuropathy was always related to risky situations of perinatal period, such as premature conditon, hyperbilirubinemia and anoxia (2,3). So, it is believed that the chances of not diagnosing hearing neuropathy by newborn hearing screening with otoacoustic emissions were minor, since it is recommended the use of otoacoustic emissions and brainstem audiometry for risky children.
Therefore, description of several cases of prelingual, in families without history of motor or sensitive peripheral neural implications (4,11), or even, cases as we have described, without neonatal or family deafness history, bring out the hypothesis of genetic origin to non-syndromic hearing neuropathy.
MADDEN et al, 2002 (12) question that before several non-syndromic family cases, a genetic predisposition to bilirubin intolerance, where lower levels to hearing loss, could cause neural change on these people.
The determination of a specific gene, when changed, that would lead to hearing neuropathy only occurred in 2003 when VARGA et al (6) identified four mutations of OTOF gene in four Spaniard families. Since then, our patients with hearing neuropathy have been submitted to mutation researchs of OTOF gene with the purpose of determining the importance and frequency of such mutation on these cases.
We found in the same patient a deletion of 16 base pairs, among nucleotides 1552-1567 (1552-1567del16). This deletion causes the appearing of a stop codon of tranlation on the next exon, which means that translated proteic product is shorter.
As it is said that mutations on OTOF gene cause deafness of recessive heredity, it is expected the patient to be a carrier of a second mutation on the same gene, which was not identified yet because sequencing study of the remaining gene is still in process. OTOF gene is a large one, with 48 exons, what makes mutation tracking process slow. Our first results show that even having analized only exons previously described as changed, there was a benefit on etiological diagnosis of deafness.
In the lilterature there are preliminary data indicating mutation frequencies of OTOF gene of 3.5% (13) among individuals with sensorineural non-syndromic hearing loss. However, this figure can be higher. As these patients might not present history that means risk to deafness and otoacoustic emissions are the method to newborn hearing screening, many cases can be left without diagnosis. The exact number determination of hearing neuropathies caused by such mutations will make us rethink of newborn hearing screening procedures.
Another point involving this diagnosis refers to the development of hearing ability from eletroacoustic devices. The results of the use of sonorous amplification device and cochlear implantation are questionable (2) and although the girl is making progress with the use of amplification, it has been slow and with difficulties.
In relation to cochlear implantation, results depend on lesion location. The closer of hearing nerve, the worse it is the implant advantage; the closer of internal ciliated cells, the better prognosis will be (12). It is impossible to determine this location before surgery and many authors have been recommended brainstem implantation when cochlear implants are not positive (14). With all these varieties of results and agreeing with the literature (12), we have chosen primarely by the use of individual sonorous amplification.
Therefore, the function of OTOF gene is to codify otoferlin enzyme which works on membrane fusion on synaptic vesicle of internal ciliated cells, providing stimulus conduction. So, patients with hearing neuropathy by mutation of this gene probably has located alteration on ICC and because of it, cochlear implantation can be more successful. Because of this diagnosis and of little advantage of hearing aid, we are sending the girl to a cochlear implantation program.
FINAL CONSIDERATIONSThe discovery of OTOF gene mutations as main cause of hearing neuropathy brings out two important clinical implications. The first is related to the diagnosis, as patients do not present visible risks to deafness, taking the chance not to be previously detected. The second is related to treatment, as lesion location is on internal ciliated cells, so recommendation of cochlear implant should be considered.
BIBLIOGRAPHY1. Star A, Picton TW, Sinninger YS, Hood LJ, Berlin CI. Auditory Neuropathy. Brain, 1996, 119(3):741-53.
2. Cone-Wesson, B, Rance G. Auditory neuropathy: a brief review. Curr Opin Otolaryngol Head Neck Surg, 2000, 8(5):421-25.
3. Martinho, ACF. Achados audiológicos em crianças com hiperbilirrubinemia neonatal: um enfoque na neuropatia auditiva. São Paulo, 2002, (Dissertação de Mestrado - Pontifícia Universidade Católica de São Paulo).
4. Wang Q, Gu R, Han D, Yang W. Familiar auditory neuropathy. Laryngoscope, 2003, 113(9):1623-1629.
5. Corley VM, Crabbe LS. Auditory neuropathy and a mitochondrial disorder in a chil: case study. J Am Acad Audiol, 1999, 10(9):484-8.
6. Varga R, Kelley PM, Keats BJ, Starr A, Leal SM, Cohn E, Kimberling WJ. Non-syndromic recessive auditory neurophaty is the result of mutations in the otoferlin (OTOF) gene. J Med Genet, 2003, 40(1):45-50.
7. Yasunaga S, Petit C. Physical map of the region surrounding the Otoferlin locus on chromosome 2p22-p23. Genomics, 2000, 66(1):110-2.
8. Yasunaga S, Grati M, Cohen-Salmon M, El-Amraouni A, Mustapha M, Salem N, El -Zir E, Loiselet J, Petit C.A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet, 1999, 21:363-369.
9. Kim TB, Isaacson B, Sivakumaran TA, Starr A, Keats BJB, Lesperance MM. A gene responsible for autossomal dominant auditory neuropathy (AUNA1) maps to 13q14-21. J Med Genet, 2004, 41(11):872-6.
10. Cone-Wesson B, Rance G, Sininger Y. Amplification and rehabilitation strategies for patients with auditory neuropathy. In: Sininger I, Starr A. Auditory Neuropathy, A new perspective on hearing disorders. 1a ed. Canadá: Singular; 2001, p.233-250.
11. Nicastro FS, Lewis DR. Neuropatia Auditiva: Estudo clínico em seis crianças. Rev Dist Comum, 2004,16(2):215-228.
12. Madden C, Hilbert L, Rutter M, Greinwald J, Choo D. Pediatric cochlear implatation in auditory neuropathy.Otol Neurootol, 2002, 23:163-8.
13. Rodríguez-Ballesteros M, Castillo FJ, Martín Y, Moreno-Pelayo M, Morera C, Prieto F et al. Auditory neuropathy in patients carrying mutation in the otoferlin gene (OTOF). Human Mutation, 2003, 22:451-6.
14. Colletti V, Carner M, Miorelli V, Guida M, Colletti L, Fiorino F. Cochlear implant failure: Is an auditory brainstem implant the answer? Acta Otolaryngol, 2004, 124:353-7.

