INTRODUCTION Hearing impairment affects 250 million people around the world and deafness is the second most common disability in developed countries(1). Loss of hair cells in mammals cause permanent sensorineural hearing loss, since these cells are terminally-differentiated, their precursors do not re-enter the cell cycle division and are held in mitotic arrest(2). Culturing cochlear cells is still a big challenge, but standardization of procedures and cell characterization are the main steps to establish the conditions for molecular, gene and stem cell therapy in the inner ear(3). The establishment of organ of Corti cultures is essential for the investigation of factors that affect hair cell and precursors cell functions and survival. The aims of this study were: 1) to establish primary cultures and subcultures from dissociated organ of Corti obtained from adult guinea pigs, and 2) to characterize these mixed cell populations with specific markers in three different culture conditions, regarding cell cycle status.
MATERIAL AND METHODS The protocol was approved by the Ethics in Research Committee of the Medical School University of São Paulo. All experiments were performed in the Otolaryngology Department and Genetic Department laboratories' of Medical School and Institute of Biosciences at the University of São Paulo and were conducted in accordance with the guidelines for the care and use of laboratory animals established by the American National Research Council.
Experimental animals Two adult guinea pigs' (Cavia porcellus) of both sexes and weighing 400 g were obtained from specialized breeders (CEDEME - UNIFESP, Sao Paulo, Brazil). The animals were housed in individual cages and given free access to food and water. Animals presenting ear infection or congenital malformations were excluded from the study. Animals were killed before the beginning of the experiments in a carbon dioxide chamber, where they remained for at least ten minutes(4).
Tissue isolation After bath in alcohol 100%, animals were decapitated; the temporal bones were removed and maintained in the Hank's balanced salt solution (HBSS). Cochlear sensory epithelia containing the organ of Corti were surgically isolated from guinea pigs' inner ears using micro-mechanical dissection technique under a stereo-microscope (Tecnival, SQF-F). The otic capsule was dissected, the epithelia containing the organ of Corti, basilar membrane, stria vascularis and spiral limbus were isolated and transferred to a flask containing 1 mL aliquot of HBSS solution (137 NaCl, 5.4 KCl, 0.3 Na
2HPO
4, 0.4 KH
2PO
4, 4.2 NaHCO
3, 5.6 glucose, 300mM HEPES at pH 7.4) and elastase 0.05 U/mL (Sigma). During enzymatic dissociation, tissue was incubated for 15 minutes at 370C, after which 3mM CaCl
2 and 600 U/mL collagenase type II (Invitrogen) were added to the aliquot for another incubation cycle. Further, 0.05% tryple (Invitrogen) was added and incubation repeated. After tissue rest, the supernatant was discarded by aspiration and the sample was washed twice with HBSS. Cells were mechanically dissociated by passing through three fine fires polished Pasteur pipettes with decreasing sizes. About 20 µL of the supernatant was added on a slide and cells morphology was observed.
Primary cell culture of the organ of Corti Cells were plated on a 35 mm Petri dish (Corning Incorporated) previously overnight covered at 370C in a 5% CO
2 incubator with 30 µg/mL poly-D-lysine (Sigma) and 2 µg/mL laminin (Sigma) and containing 2 mL of the culture medium. Cultures were maintained in a defined medium composed of DMEM-F12 (1:1, Invitrogen), 20% fetal bovine serum (FBS, Hyclone), 10 mM sodium pyruvate (Invitrogen), 1X blend solution with insulin, transferrin and selenium (Invitrogen), 2 mM glutamine (Invitrogen), 10% streptomycin and penicillin, at 370C in a 5% CO
2 incubator, and renewed every 48 hours.
Passages and establishment of subcultures The primary cell cultures were kept for two weeks until they reached 80-90% confluence, and then cells were transferred to a new culture vessel in order to propagate the population and to set up replicate cultures for cell type characterization. The subculture was performed by complete aspiration of culture medium, rinsing cell sheet twice with HBSS and adding 0.5 mL tryple (Invitrogen) on the 35 mm Petri dish (in following subcultures, bigger 100 mm Petri dishes were needed and 2 mL triple was added in these cases). Cells were maintained for 10 minutes at 370C in a 5% CO
2 incubator. Cells' attachment was monitored using an inverted microscope (Olympus CK2) to confirm their release from the substrate. Complete medium was added in a 2:1 rate (1 mL medium for each 0.5 mL tryple) to rinse cells from the flask and dislodge adherent ones. The cell suspension was then transferred to a centrifuge tube and was 6000 rpm centrifuged at 40C for 3 minutes. The supernatant was discarded; cells were re-suspended with medium and plated on new previously coated dishes.
Study Groups (control, synchronization, growth factors cultures) We carried out four passages and transferred cells into new culture dishes (35, 60 and 100 mm Petri dish) every two weeks during the first two passages, and every four days in the last two passages. In the fourth passage, cell suspension was transferred into 6-well culture plate (Corning Incorporated) containing four round coverslips per well. After 10 days of cell growth and 50% confluence, the three pairs of wells of the 6-well plate were maintained in three different study conditions regarding the cell cycle status: control, synchronization and growth factor treatment cultures.
Synchronization of culture A precise analysis of cell population types requires working with homogeneously growing cultures and synchronization of cells regarding cell cycle progression. The synchronization method allowed the arrest of the cells in the G0 phase, which was achieved by growing the cells to confluency followed by serum deprivation(5). Cells were rinsed twice with HBSS; complete medium with FBS was added in the first pair of wells of the 6-well plate and without FBS in the last four wells, and maintained at 370C in a 5% CO
2 incubator for 24 hours. The pair of wells without FBS received 20 ng/mL epidermal growth factor (EGF) and 10 ng/mL beta 2 fibroblast growth factor (
b-2-FGF), and cells were maintained at 370C in a 5% CO
2 incubator for 24 hours.
Phenotypic cellular characterization using specific markers and immunofluorescence Cells in the coverslips (from wells kept under three different conditions) were fixed for 20 minutes in methanol at minus 200C for optimal penetration of IgM antibodies into nuclei, rinsed in HBSS, permeabilized in 0,3% Triton X-100 for 20 minutes at room temperature. Cells were blocked with 10% goat serum (Santa Cruz Biotechnologies) and incubated with primary antibodies diluted with 3% bovine serum albumin (BSA, Gibco) and HBSS for one hour at room temperature, according to manufacturer's instructions: beta 3 tubulin (1:250 mouse; 1µL antibody + 250µL BSA 0.3% diluted in HBSS), nestin (1:250 mouse; 1µL antibody + 250µL BSA 0.3% diluted in HBSS), myosin VIIa (1:25 rabbit; 4µL antibody + 100µL BSA 0.3% diluted in HBSS), connexin 26 (1:500 mouse; 0.5µL antibody + 250µL BSA 0.3% diluted in HBSS). Cells were rinsed in HBSS and incubated with secondary anti-species specific antibodies diluted in BSA and HBSS for one hour at room temperature: anti-rabbit FITC (1:200, 1 µL antibody + 200µL BSA 0.3% diluted in HBSS) for myosin VIIa, anti-mouse Cy3 (1:200, 1µL antibody + 200µL BSA 0.3% diluted in HBSS) for beta 3 tubulin, nestin and connexin 26. Cells in the coverslips were mounted in vectashield system (Vector Laboratories, CA, USA) containing DAPI for nuclear identification and sealed with nail polish. Cells were examined using a fluorescence microscope (ZEISS Axioplan) equipped with chroma filters and a software used to collect digital images (Isis Fish Imaging Meta System).
Quantitative analysis of immunofluorescence results Quantitative analysis was performed through direct counting of the number of cells labeled with the specific antibodies (beta 3 tubulin, nestin, myosin VIIa, and connexin 26) and division by the total number of cells corresponding to DAPI nuclear positivity, from 20 randomly chosen microscope fields, 40X objective magnifying.
Statistical Analysis The results were expressed as the mean ± standard deviation of the percentage of labeled cells per coverslip, in each group (control, synchronization, growth factor). The continuous variables were compared by one-way analysis of variance (ANOVA). The level of statistical significance was set at p
< 0.05. Statistical analysis was performed using the GraphPad Instat program.
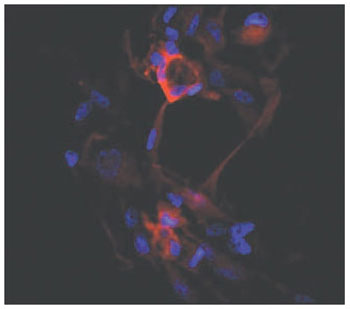
RESULTS The number and percentage of labeled cells per coverslip is presented in Table 1 (descriptive analysis). There were no cells stained after anti-nestin treatment. Nestin is an intermediate filament protein, mainly expressed in immature neurons. Few cells showed labeling after anti-
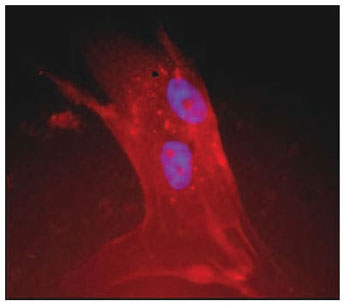
b-3-tubulin staining (11.2%), which is a specific mature neuronal marker. The greater proportions of stained cells were found after anti-f -myosin VIIa (51.5%) and anti-connexin 26 treatments (46.5%), which are specific markers for hair cells and support cells, respectively. Comparisons of the mean and standard deviation of the labeled cells between groups are presented in Table 2. Although only few cells stained with anti-
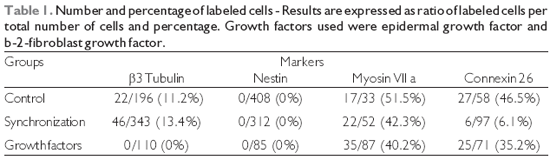
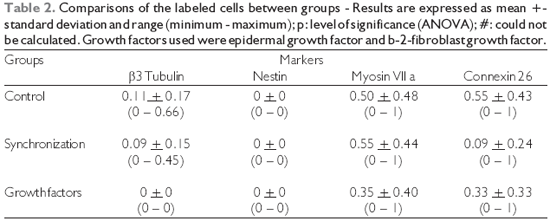
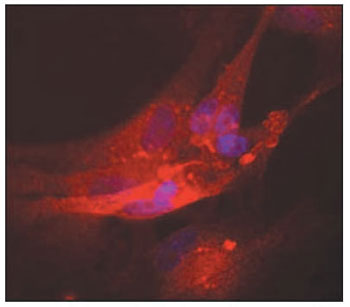
b-3-tubulin, there was a significant difference between groups, as no labeling was observed in the cultures treated with growth factors (p = 0.013, ANOVA). The level of significance could not be calculated for the anti-nestin labeling experiment since there was no staining at all. Regarding anti-myosin VIIa staining, there was no significant difference between groups (p = 0.35, ANOVA). A significant difference was observed for anti-connexin 26 staining between groups (p = 0.0004, ANOVA), as there was a striking reduced proportion of stained cells among those submitted to synchronization (mean = 0.09) in comparison with the control experiment (mean = 0.55). A trend for increased proportion of stained cells was observed after growth factor treatment, but the proportion was smaller than the control (mean = 0.33). The immunofluorescence images of the organ of Corti cultures could be obtained for anti-
b-3-tubulin, anti-myosin VIIa and anti-connexin 26 staining and the results are presented in figures 1, 2, and 3, respectively.
DISCUSSIONThe immunofluorescence studies confirmed the epithelial phenotype of the cell cultures primary collected from the organ of Corti after micro-mechanical dissection and maintained through a few passages. The cells were mainly positive for staining with antibodies that detect epithelial cell markers (myosin VIIa and connexin 26) and negative or weakly stained with antibodies for neuronal markers (nestin and
b-3-tubulin). Our results agree with others in the literature, although the culture methods we employed are different(3, 6, 7). Although we analyzed cells with only 50% confluence, which could suggest a low frequency of intercellular junctions, we did observe connexin 26 staining. This could be explained by the fact that this protein was also reported as having perinuclear localization(8). The synchronization is a useful technique to standardize cell growth in different culture plates and to drive them into the same point of the cell cycle(5). However, synchronization led to a decrease in cell number. The growth factor treatment favored the selection of epithelial cells in the culture coverslips, since mature and immature neuron markers were not detected in these experiments (Table 1). Growth factor treatment seems to be important in cell selection, and in the near future such strategy will probably be applied in molecular, gene and stem cell therapy in hair cell and inner cell regeneration(2). Summing up, primary cultures and subcultures derived from dissociated organ of Corti and cochlear tissue were established and have yielded cell types, which comprised few mature neurons but mainly epithelial cells, as hair cells and support cells.
CONCLUSIONS Primary cultures and subcultures derived from dissociated organ of Corti were established and have yielded cell types, including few mature neurons but mainly hair cells and support cells. The synchronization procedure led to a decrease in the number of epithelial cells obtained. The treatment with growth factors was essential for selecting epithelial cells.
REFERENCES1. World, Health, Organization. www.who.int/pbd/deafness/en. In; 2007.
2. Taylor R, Forge A. Developmental biology. Life after deaf for hair cells? Science 2005;307(5712):1056-8.
3. Zhao HB. Long-term natural culture of cochlear sensory epithelia of guinea pigs. Neurosci Lett 2001;315(1-2):73-6.
4. Souza NL. Eutanásia. In: Luca RR, Alexandre SR, Marques T, Souza NL, Merusse JLB, Neves SP, editors. Manual para técnicos em bioterismo. 2a ed. São Paulo: FINEP-COBEA; 1996. p. 157-177.
5. Schorl C, Sedivy JM. Analysis of cell cycle phases and progression in cultured mammalian cells. Methods 2007;41(2):143-50.
6. Malgrange B, Belachew S, Thiry M, Nguyen L, Rogister B, Alvarez ML, et al. Proliferative generation of mammalian auditory hair cells in culture. Mech Dev 2002;112(1-2):79-88.
7. Li H, Liu H, Heller S. Pluripotent stem cells from the adult mouse inner ear. Nat Med 2003;9(10):1293-9.
8. Marziano NK, Casalotti SO, Portelli AE, Becker DL, Forge A. Mutations in the gene for connexin 26 (GJB2) that cause hearing loss have a dominant negative effect on connexin 30. Hum Mol Genet 2003;12(8):805-12.
1. MD PHD (OTOLARYNGOLOGY DEPARTMENT UNIVERSITY OF SÃO PAULO MEDICAL SCHOOL)
2. MD PRECEPTOR (OTOLARYNGOLOGY DEPARTMENT UNIVERSITY OF SÃO PAULO MEDICAL SCHOOL)
3. PHARMACY MS (INSTITUTE OF BIOSCIENCES GENETIC DEPARTMENT UNIVERSITY OF SÃO PAULO)
4. BIOLOGY PHD (INSTITUTE OF BIOSCIENCES GENETIC DEPARTMENT UNIVERSITY OF SÃO PAULO)
5. MD PHD (INSTITUTE OF BIOSCIENCES GENETIC DEPARTMENT UNIVERSITY OF SÃO PAULO)
6. MD PHD (PROFESSOR AND CHAIRMAM OTOLARYNGOLOGY DEPARTMENT UNIVERSITY OF SÃO PAULO MEDICAL SCHOOL)
OTOLARYNGOLOGY DEPARTMENT, UNIVERSITY OF SÃO PAULO MEDICAL SCHOOL, SÃO PAULO, BRAZIL / INSTITUTE OF BIOSCIENCES, UNIVERSITY OF SÃO PAULO, SÃO PAULO, BRAZIL
JEANNE OITICICA, MD PHD
Av. Damasceno Vieira, 1202 ap. 41 - Vila Mascote - CEP: 04363-040 - SÃO PAULO/SP, BRASIL - FONE(FAX): +55(11) 5671-6810 / 7436-1596 - Email: jeanneoiticica@bioear.com.br
Este artigo foi submetido no SGP (Sistema de Gestão de Publicações) da R@IO em 7/10/2007 e aprovado em 10/10/2007 16:23:12.