INTRODUCTION The nasal obstruction is potentially severe when it affects the newborns, since they are the preferential nasal breathers. Such characteristic remains on average until the fifth month of life (1,2). In this age range, the tongue takes practically all the space of the oral cavity, and remains along the palate, the larynx is at a higher position and the epiglottis reaches or even outreaches the soft palate towards the nasopharynx. Therefore, the upper airways anatomy allows the maintenance of breathing by nasal way and deglutition at the same time (2). Then the newborns with nasal obstruction generally presents with feeding difficulty, cyanosis, choking or need for pauses for breathing during breastfeeding. In addition to the symptoms associated with feeding, many infants have cyclical cyanosis that improves with crying because in this condition the newborn starts oral breathing. Despite the most common cause of nasal obstruction of the newborn and the infant is the mucous edema secondary to viral rhinitis or idiopathic rhinitis of the infant (3), it is important to be attentive to the diagnosis of nasal anatomic anomalies. The objective of this series of cases is to present three patients who represent the most common causes of anatomic nasal obstruction in the newborn and alert for the importance of diagnostic research and early management of such cases.
CASES REPORT Case 1
Female patient, born of vaginal birth without complications, presented breathing dysfunction right after the birth, with difficulty of progression of the aspiration probe in both nasal cavities. The newborn was intubated in the birth room and forwarded to the Neonatal Intensive Care Unit (NICU). Two extubation attempts were carried out, but the patient had a growing breathing difficulty, removal of wishbone and intercostal membrane and intermittent cyanosis (relieved by crying), and new oral intubation was necessary. On the sixth day of life, the otorhinolaryngology evaluation was requested. The basic otorhinolaryngological exam was normal and upon fibronasoendoscopy we observed bilateral choanal atresia. Computed Tomography of the facial bones was requested, which confirmed bilateral atresia with a significant posterior thickening of the vomer (Picture 1). The genetic evaluation discarded other malformations. On the eighth day of life, the patient was submitted to endoscopic correction of the choanal atresia, and silicon stent was used for four days. In the postoperative period, there were no complications and hospital discharge occurred on the seventh day, without symptoms. Three months after surgery, the patient had obstruction symptoms again and we confirmed the full closure of the choana to the right and stenosis to the left. She was again submitted to endoscopic opening, this time without use of stent in the postoperative. After the second procedure, the evolution was excellent, with discharge on the third postoperative day. Currently the patient is aged 18 months and has no symptoms.
Case 2
Female, white patient aged 13 days of life, with exclusive breastfeeding, was transferred to a tertiary hospital for presenting with tachypnea, breathing noise and ventilatory effort. Her mother reported the clinical picture was present since birth, with a worsening during breastfeeding. She also reported many pauses during feeding. The patient was born with 35 weeks of gestational age, by vaginal birth, and weighing 2980 grams. There were no complications during pregnancy, except for preeclamptic toxaemia in the peri-birth. Upon examination, the newborn presented tachypnea, with severe inspiratory noise and extracostal air. There was difficulty in the nasal progression of the probe of 3 mm. We also identified systolic murmur and presence of genitourinary fistula. The complimentary exams revealed iron deficiency anemia, echocardiogram without alterations, echography of the urinary canal with pielocalycial dilation to the right, normal karyotype and altered electroencephalogram, requested for episode of extreme tremors. With suspicion of choanal atresia, CT of the facial bones was requested, which presented an image compatible with bilateral nasolacrimal duct and pervial choanas (Picture 2). During nasal endoscopy, it was difficult to pass the flexible endoscope of 3.4 mm and we identified a cystic lesion at the level of the inferior meatus on bilateral position. During this diagnostic research, the newborn presented a progressive improvement of the breathing dysfunction and oral feeding. We then opted for the conservative procedure. In the service follow up, the patient, now aged 8 months, evolves without respiratory dysfunction and has a normal diet acceptance.
Case 3
Male newborn presented with respiratory dysfunction for nasal obstruction in the first hours of life and otorhinolaryngologic evaluation was requested. The patient was born on time, weighing 3260g, of vaginal birth without complications, APGAR 9 and 10. In the evaluation, he had inspiratory noise, nasal obstruction and intermittent cyanosis with relief when crying. There was a report of difficulty with progression of nasal probe and it was only possible to progress aspiration probe number 2 at the right. Upon anterior rhinoscopy, a diminishment of the space of the nasal valve was observed and it was not possible to proceed with fibronasolaryngoscope of 3.4mm in both nasal cavities. CT was requested for the facial bones. In this exam we confirmed congenital stenosis of anterior pyriform aperture with a space of 0.07cm to the left and 0.10cm to the right (Picture 3), in addition to the solitary maxillary central incisor. Initially, the conservative treatment was attempted with nasal washing, but, because the patient kept on presenting cyanosis crisis and difficulty to breastfeed, the surgical procedure was indicated. It was carried out on the sixth day of life, through degloving access), with drilling of the maxilla medial nasal process. The patient was forwarded to the NICU with silicon tubes number 3.5mm which molded the nasal cavities and orally intubated (Picture 4). He was extubated in the first 24 hours and the moulds were removed on the third postoperative day. The patient restarted breastfeeding on the fourth postoperative day, without complications. The nasal breathing was taken over, there was no more episodes of cyanosis, and the patient was discharged on the fifth postoperative day. Now the patient has been followed up for 1 year with a good development and without nasal obstruction.
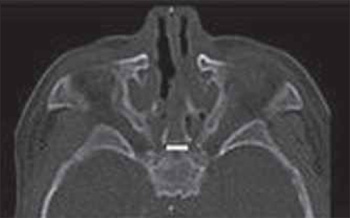
Picture 1. Tomographic cut demonstrating bilateral choana atresia.
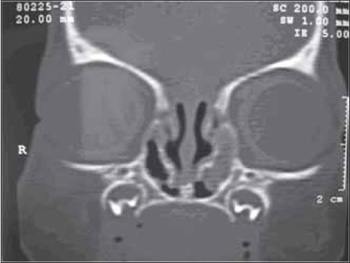
Picture 2. Tomographic coronal cut demonstrating bilateral nasolacrimal duct mucocele. Mucocele to the left presents a filling with material with soft parts density, probably associated with secretion.
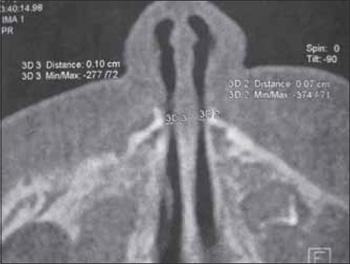
Picture 3. Tomographic axial cut demonstrating bilateral pyriform aperture stenosis.
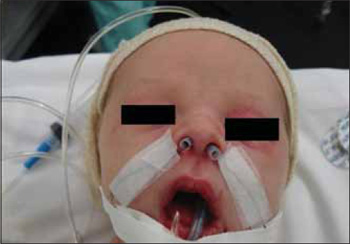
Picture 4. Aspect of the patient in the immediate postoperative, with silicon moulds in the nasal cavities and orotracheal tube.
We present three cases of nasal obstruction manifested in the first days of life caused by different pathologies, but with a clinical picture practically indistinguishable. Because the nasal anatomic alteration is more frequent, the choanal atresia is generally the first hypothesis.
The exuberance of clinical manifestations will depend upon the unilateral or bilateral position and the level of obstruction. The classical clinical picture in bilateral cases of severe obstruction is of a ventilatory effort, tachypnea and subsequent cyanosis. The absence of shaking of the nasal wing in a case of breathing dysfunction suggests the diagnosis of nasal obstruction. The generated hypoxia unchains the crying and when it starts the obstruction is relieved because it allows oral breathing. When the oxygenation improves and the crying ceases a new picture of obstruction is started, followed of hypoxia and crying and so on. The children who acquire oral breathing and outreach this phase without treatment may present with feeding difficulties for they cannot suck suitably. However, as described by WILLIAMS in 1971 (4), there are some patients who quickly "learn" the oral breathing, which for instance explains uncommon cases of bilateral choanal atresia diagnosed in the adult life.
In cases of unilateral obstruction, the diagnosis may be made by the disability to progress with an aspiration probe through the affected nasal cavity. But in many cases the diagnosis is late, when the patient seeks medical service for long time ago unilateral nasal obstruction, anosmia and rhinorrhea.
As in every non frequent pathology, the high degree of suspicion is basic for the diagnosis of the nasal congenital anomalies. As already described, the first form of diagnosis is the nonprogression of a probe by the nasal cavity. Once the suspicion is raised, the next step is to obtain a pervious airway, and it is necessary to proceed with the placement of an oral cannula (Guedel tube or McGovern's dummy) or even oral intubation. The otorhinolaryngologist's evaluation must be requested as soon as there is a diagnostic suspicion and includes anterior and posterior rhinoscopy through nasoendoscopy. The imaging exams are critical for the diagnostic definition and the possible surgical planning.
The choanal atresia is the most frequent congenital nasal anomaly. It has an incidence of 1: 4000 to 1: 10000 living newborns and is more common in girls (5.6). It represents a malformation of the posterior nasal opening that impairs the air passage into the rhinopharynx. It may be unilateral or bilateral, and is unilateral in 60-70% of the cases (5). In terms of composition, it may be classified as osseo-membranous (two-thirds of the cases) or purely osseous (7). Separate or part of a syndromic picture, and its association has already been described with more than 20 syndromes. The most frequent syndromic picture is represented by the acronym CHARGE (coloboma, congenital heart defect, atretic choanae, retarded physical and neuromotor development associated with CNS anomalies, genital hypoplasia, ear anomaly and/or deafness), and choanal atresia is present in 60% of the cases (8). The choanal atresia was also described after exposure to methymazole during pregnancy (8). Therefore, we suggest geneticist's evaluation of all patients with choanal atresia to exclude other possible malformations. In the case described the choanal atresia was an isolated malformation. The imaging exams are essential for the diagnosis, and CT is the gold standard exam. The choanography (lateral X-Ray after placement of contrast in the nasal cavities), in spite of not responding to important questions for the surgical planning, has an essential diagnostic value where the radiologic resources are limited. The treatment for choanal atresia is always surgical, and the intensity of the clinical manifestations determines the intervention moment. In the newborn presented, the treatment was early due to the severe clinical picture manifested. The sooner the patient needs intervention the higher the probability of restenosis with need for more than one surgery for maintenance of the pervious choana. The access to the choanal opening may be transnasal or transpalatine. With the arrival of microscopy and endoscopy, the transnasal passage is the most accessible for the shortest surgical time, less bleeding risk, low morbidity and minimal risk of lesions and structures in development (5).
The congenital dacryocystocele was first described by RAFLO in 1982 as an obstruction of the nasolacrimal system and with an uncommon cause of respiratory dysfunction of the newborns, when expanded to the nasal cavity (9). There is no clear differentiation in the literature between the terms dacryocystocele and nasolacrimal duct cyst, and they are even applied as synonyms (10). We use the term dacryocystocele both for distal and proximal obstruction in the nasolacrimal system, according to TSAI (11). This obstruction occurs in more than 80% of the newborns. However, only in 2 to 4% of the cases this obstruction becomes symptomatic and in 80% of these, the spontaneous rupture occurs around the sixth month of life. The congenital dacryocystocele is more common in the Hispanic people and is from 3 to 9 times more common in the female sex, which has been ascribed to a narrower nasolacrimal duct in this group (3, 10). It may clinically appear upon birth or in the first weeks of life as a medial canthal or asymptomatic nasal mass, epyfore, dacryocystitis, periorbitary cellulitis, sepsis, nasal obstruction and seldom respiratory dysfunction (12). In the literature there is no description of associated anomalies (8), differently from the patient presented, who had genitourinary and neurological alteration. The diagnosis is made through the clinical history, nasal endoscopy and imaging exam. The endoscopy may show a cystic mass on the level of the lower meatus, as in the case reported. The nuclear magnetic resonance (NMR) is superior to CT in the nasolacrimal duct study, but the imaging exam is also requested to evaluate atresia or stenosis of choanas and other nasal abnormalities, which makes CT the choice exam for the research of nasal obstruction in the newborn (9, 12). The treatment of dacryocystocele may be conservative in the absence of respiratory commitment and includes massage, nasal washing, hot compresses and catheterization of the nasolacrimal duct (3, 10, 12). The spontaneous resolution generally occurs in 85-95% of the cases in the 12 first months of life (12) and this was the evolution observed in the patient described. When there is respiratory commitment that interferes with feeding or when the parents are not very confident to maintain the child at home with nasal obstruction, the surgical intervention may be adequate, and endoscopic marsupialization of the cyst is the treatment recommended (9, 10).
First described by BROWN in1989 (13), the congenital nasal pyriform aperture stenosis (CNPA) is also an uncommon cause of nasal obstruction of the newborn. It is characterized by a narrowing of the nasal cavity (at the beginning of the osseous part) for the excessive growth of the maxilla medial nasal process (14). It is taken as a minor form of holoprosencephaly, a failure of the development of the prosencephalon and the medial facial structures. Such hypothesis is based on the presence of the solitary maxillary central incisor, which is one of the manifestations of the holoprosencephaly and is present in half of the cases of CNPA (13, 15, 16). The patient reported presented with this alteration. Therefore, despite the CNPA and the presence of the solitary maxillary central incisor can be separate findings, the pituitary hypoplasia and other abnormalities of the central nervous system need to be excluded (8), preferably with NMR (15). The CNPA is generally bilateral and manifests upon birth. The CT with parallel cuts of the hard palate is the choice imaging exam. A pyriform aperture is considered to be stenotic when the transversal diameter of each aperture is lower or equal to 3 mm or the total transversal diameter of the pyriform aperture is lower than 8-11mm in a normal newborn (8.15). The reported patient presented 0.7 mm to the left and 1.0mm to the right of transversal diameter, which confirms the diagnosis of CNPA. If the patients obtain a good breathing and adequate dietary ingestion with nasal washing, they can be clinically managed, once the symptomatic improvement generally occurs at about 6 months of age. The surgical enlargement of the pyriform aperture is recommended in those patients with breathing and feeding difficulty (16). The access may be transnasal (technically difficult in the nose of a newborn) or sublabial, with a better viewing and preservation of the nasal mucosa, the technique used in the case reported. The bone of the aperture must be worn enough to allow the passage of an endotracheal tube of 3.5mm.
CONCLUSION The newborn's nasal obstruction may appear in a dramatic manner, which causes severe respiratory dysfunction and deglutition disorders with secondary aspiration. The detailed evaluation of these pictures is essential for the early diagnosis and adequate management, aiming to prevent complications. The difficulty of the nasal way aspiration probe progression in the routine exam in the birth room must alert the neonatologist as for the anatomic obstruction of the nasal cavity, specially if the newborn presents with clinical respiratory or deglutition difficulty. The pictures of choanas atresia, dacryocystocele and congenital stenosis of the pyriform aperture may be clinically indistinguishable, but they present distinct therapeutic approaches and the suitable differential diagnosis is fundamental.
BIBLIOGRAPHIC REFERENCES 1. Faust RA, Phillips CD. Assessment of congenital bony nasal obstruction by 3-dimensional CT volume rendering. Internacional Journal of Pediatric Otorhinolaryngology. 2001, 61:71-5.
2. Endo LH, Montenegro MCS. Obstrução nasal no recém nascido e na criança. Tratado de otorrinolaringologia. Volume 3; 1 edição; 2003, 175-80.
3. Brachlow A, Schwartz RH, Bahadori. Intranasal mucocele of the nasolacrimal duct: an important cause of neonatal nasal obstruction. Clinical Pediatrics. 2004, 43:479-81.
4. Williams HJ. Posterior choanal atresia. Am J Roentg. 1971, 112(1):1-11.
5. Kuhl G, Smith MM, Monteiro FM. Atresia coanal. In: Costa SS, Cruz OLM, Oliveira JA. Otorrinolaringologia Princípios e Prática 2a edição. Porto Alegre: Artmed; 2006. p. 735-42.
6. Hengerer AS, Brickman TM, Jeyakumar A. Choanal Atresia: Embryologic Analysis and Evolution of Treatment, a 30-Year Experience. Laryngoscope. 2008, 118:1-5.
7. Brown OE, Pownell P, Manning SC. Choanal Atresia: a New Anatomic Classification and Clinical Management Applications. Laryngoscope. 1996, 106(1):97-101.
8. Vanzieleghem BD, Lemmerling MM, Vermeersch HF, Govaert P, Dhooge I, Meire F et al. Imaging Studies in the Diagnostic Workup of Neonatal Nasal Obstruction. Journal of Computer Assisted Tomography. 2001, 25(4):540-49.
9. Calcaterra VE, Annino DJ, Carter BL, Woog JJ. Congenital nasolacrimal duct cysts with nasal obstruction. Otolaryngology Head and Neck Surgery. 1995, 113(4):481-4.
10. Hepler KM, Woodson GE, Kearns DB. Respiratory Distress in the neonate: sequela of a congenital dacryocystocele. Archives of otolaryngology-head and neck surgery. 1995, 121(12):1423-5.
11. Tsai YS, Huang JK. Neonatal nasal obstruction caused by bilateral dacryocystoceles. Pediatr Radiol. 2006, 36:1221.
12. Roy D, Guevara N, Santini J, Castillo L. Endoscopic marsupializaton of congenital nasolacrimal duct cyst with dacryocoele. Clin Otolaryngol. 2002, 27:167-70.
13. Brown OE, Myer CM, Manning SC. Congenital nasal pyriform aperture stenosis. Laryngoscope. 1989, 99:86-91.
14. Tagliarini JV, Nakajima V, Castilho EC. Congenital nasal pyriform aperture stenosis. Rev Bras Otorrinolaring. 2005, 71(2):246-249.
15. Rollins N, Booth T, Biavati M. Case 40: Congenital Pyriform Aperture Stenosis. Radiology. 2001, 221(2):392-4.
16. Lee JJ, Bent JP, Ward RF. Congenital nasal pyriform aperture stenosis: non-surgical management and long-term analysis. International Journal of Pediatric Otorhinolaryngology. 2001, 60:167-71.
1. Resident Doctor of the Otorhinolaryngology Service of the Clinical Hospital of Porto Alegre.
2. Contractor Doctor of the Otorhinolaryngology Service of the Clinical Hospital of Porto Alegre. Master's Degree in Pediatrics at the Federal University of Rio Grande do Sul.
3. Fellow Doctor in Pediatric Laryngology and Airways of the Otorhinolaryngology Service of the Clinical Hospital of Porto Alegre.
4. Professor of the Ophthalmology and Otorhinolaryngology Department of the Medical School of the Federal University of Rio Grande do Sul.
Institution: Hospital de Clínicas de Porto Alegre. Porto Alegre / RS - Brazil. Mail Address: Denise Manica Avenida - João Pessoa, 1051/408 - Bairro Cidade Baixa - Porto Alegre / RS - Brazil - Zip code: 90040-000 - Telephone: (+55 11) 2101-8249 - E-mail: denisemanica@gmail.com
Article received on July 27 2008. Article accepted on April 5 2009.

