 |
657 |
|
| Year: 2009 Vol. 13 Num. 4 - Out/Dez - (14º)
|
|
 |
|
|
| Langerhans Cell Histiocytosis in Otorhinolaryngology |
|
| Author(s): |
| Humberto de Barros Fernandes1, Ronaldo Campos Granjeiro2, Jacinto de Negreiros Júnior3.
|
|
|
| Key words: |
| Langerhans cell histiocytosis, otorhinolaryngology, temporal bone. |
|
|
 |
| Abstract: |
Introduction: The Langerhans cell histiocytosis is an inflammatory cells proliferative disorder of unknown etiology. It is uncommon disease in children. Objective: To proceed with a literature review on Langerhans cell histiocytosis, with focus on the otorhinolaryngological complications. Materials and methods: The methodology used was advised based on online data from MEDLINE, between 1966 and 2008, with research of terms related to Langerhans cell histiocytosis, temporal bones and otorhinolaryngology. Literature Review: The manifestations in the head and neck are the most common ones and their diagnosis becomes difficult once it mimetizing other more common diseases the otorhinolaryngologist sees as external ear eczema, acute mastoiditis and gingivitis. The temporal bone disease manifests as recurrent otorrhea and external auditory meatus and retroauricular granulomas. The radiological evaluation confirms lytic lesions especially in the cranial cap, jaw, temporal bones and spines. The definitive diagnosis is made by biopsy through the histopathological discoveries and immunohistochemistry detection of the CD1a antigen. The main form of treatment is by chemotherapy and, in a lower scale, radiotherapy or surgery. Conclusions: he otorhinolaryngological manifestations must be suspected for recurrent otological symptoms and the presence of retroauricular granulation tissue or and the external auditory meatus. The biopsy with characteristic histological discoveries and immunohistochemistry positive for CD1a were diagnostic. The chemotherapy may be the initial treatment in most cases or secondary in refractory or recurrent forms.
|
|
|
INTRODUCTION
The Langerhans cell histiocytosis (LCH) is a disease of the histolytic disorders group, whose primary event is the accumulation and infiltration of monocytes, macrophages and dendritic cells in the tissues affected. This description excludes diseases in which the infiltration by such cells occurs as a response to a primary pathology (1). The Langerhans cells, strictu sensu, correspond to the defense cells present in the skin. The term is included in the name of the disease because the cells that proliferate - histiocytes, also called dendritic cells - are phenotypically similar to the Langerhans cells of the skin.
The first report of the disease was made in 1865, when THOMAS SMITH described the case of a child with impetigo and osseous lesions in the cranium (1). Since then, several reports of children with a picture of exophthalmia, osseous lesions and diabetes insipidus have appeared, amongst which those described by HAND, SCHÜLLER and CHRISTIAN, who named one of the forms of the disease (2). In 1924, LETTERER and SIWE described a more severe and lethal form of the visceral disease which consisted of cutaneous lesions, hepatosplenomegaly, lymphadenopathy and pneumonia (1,2). In 1953, LICHTENSTEIN gathered the several clinical forms under the name histiocytosis X and later, in 1973, NEZELOF modified it to Langerhans cell histiocytosis (1).
As for the etiology, there are works that appoint a monoclonal neoplastic disorder (3), and it is not yet known whether it results from a genetic defect or an abnormal response to the infection, trauma or autoimmune phenomenon. We verified the main cytokines related to the blood formation and the inflammatory processes are produced mainly by the histiocytes, lymphocytes and macrophages (4) This indicates that once the activation of these cells is started, they perform a self-amplification of their stimulus, which progresses with the disease.
LITERATURE REVIEW
Materials and Methods
The literature review was carried out through the on-line database Medline, by using the terms Langerhans cell histiocytosis, otorhinolaryngology and temporal bone for research, in the period between 1966 and 2008.
Epidemiology
The incidence of the LCH is estimated in about 3-5 cases per million of children; it is possible that this data is underestimated, since there are non-diagnosed osseous and cutaneous lesions. In newborns this value is of 1-2 per million. As for sex, there is a slight predominance of male sex at an overall proportion of 1.5:1. In the patients with unifocal affection this proportion falls to 1.3:1, while in the multisystemic cases it remains at 1.9:1 (5). The age range of the disease is mainly pediatric, although in the literature there are reports of the disease in adults (6,7). The incidence peak is from 1 to 3 years of age and the patients with focal lesions are generally older (0.1-15.1 years) than those with multisystemic disease (0.09-14.8 years) (5).
Classification
The histiocytes diseases classification is difficult and the systems that propose to proceed with it comprise a large number of diseases. The classical disease division into categories of eosinophilic granuloma, Hand-Schüller-Christian's disease and Letterer-Siwe's disease are still in the International Classification of Diseases (CID10), but in different regions: the latter is included in the malignant diseases and the first two are included in the benign hematological diseases. Today, the most utilized classifications are those by the World Health Organization (WHO) and the Histiocyte Society. The following is that of CID10 (8) (Graphic 1).
The classification of histiocytosis by the Society was made in 1997 and is based on the group of cells present in the lesions (9) (Graphic 2).
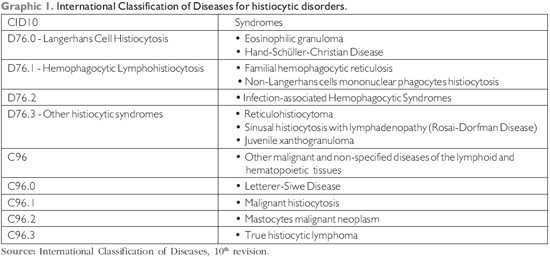
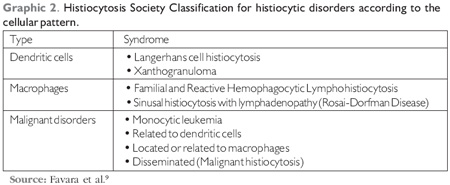
Clinical Manifestations
The LCH may have a much diversified clinical picture, since asymptomatic - as in some isolated osseous lesions - up to severe manifestations with lethal perspective. Most of which start the disease with signs and symptoms in the head and neck in up to 82% of the cases and along the disease the frequency of such symptoms increase. The most frequent is the osseous affection (78% of the patients), more common in the cranial cap, followed by the femur, orbit and spine (2). O diabetes insipidus is the most frequent endocrinopathy in the LCH, with reports of up to 50% of the cases. There may be maxillomandibular affection and gingival disease leading to the loss of dental elements, ulceration, mucous bleeding or early dental eruption in very young children (2).
The temporal bone affection by histiocytosis occurs in 15 to 60% of the cases and is higher when radiological findings are included in asymptomatic patients; its bilateral occurrence is described in up to 30% of the cases (9, 10) and is more frequent in the multisystemic disease (2, 11); in 5 to 25% of the cases, the otological signs and symptoms may be the initial clinical manifestation (12). These manifestations confuse and retard the diagnosis, because these patients initially seek an otorhinolaryngologist and the first hypothesis includes the infectious etiologies - an important cause of otorrhea in the childhood. The most frequent symptoms are recurrent purulent otorrhea and retroauricular edema in the mastoid. The polyps of the meatus are also frequent discoveries and their dissemination occurs through the posterior wall of the meatus. Because the disease is predominant in children, the hearing loss is not a much reported symptom, but when tested we observe several levels of conductive loss. The inner ear lesions are seldom, which shows the optical capsule is highly resistant to invasion by a granulomatous tissue (13).
Cutaneous lesions are verified in up to 50% of the patients, and may be the only sign of the disease or evidence of multisystemic commitment. One of the most common forms is the rash, in addition to the cutaneous infiltrates in the maculoerythematous form, in the form of xanthomatous petechias, nodes or papules. The preferential location is the middle line of the trunk and the flexor areas of the limbs. The scalp has erythematous lesions that may evolve into petechias that ulcerate and form crusts; these are not pruriginous lesions and there may be alopecia. In children of one year of age there is a nodular form of the disease remarkable for the lesions eruption similar to varicella, which is called self-limiting histiocytosis, because it has spontaneous improvement (14).
The pulmonary involvement is observed in 20-40% of the patients and manifests clinically with cough, tachypnea, dyspnea or even pneumothorax. In addition to this, the radiological images show a micronodular or cystic infiltrate. The lung function tests reveal a restrictive pulmonary disease and a reduced pulmonary volume (15). The main complication of the gastrointestinal lesions is bleeding; in the hepatic involvement the aminotransferases and, less commonly, the bilirubins increase. The hematological alterations may mean a lesion in the bone marrow or in the spleen. The lymph node infarction is verified in about 30% of the cases. They are seldom symptomatic. When symptomatic they may obstruct or damage adjacent structures, when they affect the respiratory tract, they cause cough, dyspnea and cyanosis and when superficial they lead to suppuration (15).
Differential Diagnosis
Diagnosing the LCH depends upon a high level of suspicion, since its clinical manifestation is highly diversified. As for the bone lesions, the malignant diseases (metastases), neuroblastomas, primary osseous sarcomas and even leukemia must be clinically debriefed. By adding the radiological findings, other diseases begin to be included such as meningioma, hemangioma, rabdomiossarcoma, congenital cholesteatoma, dermoid or arachnoid cysts, meningoencephaloceles, arteriovenous malformations, fibrous dysplasia, hyperthyroidism and osteoradionecrosis. Because these patients may present with chronic inflammatory signs, we must also think of osteomyelitis and tuberculosis (16). The cutaneous lesions must be differentiated form other dermatological lesions such as napkin rash, seborrheic dermatitis, In patients with the disease manifested as ganglion infarction we must discard lymphomas, malignant histiocytosis, metastases, spindle cell nevus, mastocytosis, infectious and granulomatous diseases. The otorrhea must amplify the diagnosis into acute suppurative otitis media, otomastoiditis and chronic otitis (13, 17). A chronic otological disease of critical suspicion would be the congenital cholesteatoma, especially in the presence of dysacusis. Since both these entities are very similar clinically, a parameter often mentioned to differentiate them is the erythrocyte sedimentation rate that is increased in the histiocytosis (18). Oral lesions must be told from the gingivostomatitis. In patients with pulmonary, hepatic and gastrointestinal complications, we must research immunodeficiency, leukemia and solid tumors metastases (15). The diabetes insipidus must have its cause investigated, because in addition to the HCL, there is hypothalamic-hypofisary affection to the ademonas, craniopharyngiomas, sellar chondromas, meningiomas, gangliocytomas, optical and hypothalamic gliomas and germinative cells tumors. Genetic syndromes with chromosomal disorders such as multiple endocrine neoplasia type I (MEN1), familial acromegaly, McCune-Albright syndrome and Carney syndrome may also be associated with the hypofisary tumors (19).
Diagnosis
Based on the clinical suspicion, after biopsy an anatomopathological study is carried out and the diagnosis is established. Despite the clinical variations, the LCH histology is generally uniform and does not seem to relate to the prognosis of the disease. The discoveries may have some differences according to the time and region of the disease. In early lesions there are pathologic Langerhans cells, macrophages, interdigital cells, lymphocytes T and giant histiocytes; there may also be eosinophiles and necrotic cells. Over time, the cellularity diminishes and the macrophages increase, as well as the fibrosis (20). From a morphologic viewpoint, the Langerhans cells have about 12 m of diameter with cytoplasm at a moderate quantity, as well as a lobate and a wheel-spoke nucleus with one to three nucleoli and a prolonged central fissure that gives it the appearance of a bean. In the LCH, the cytoplasm and seldom the nucleus contain the characteristic structures called Birbeck granules that help the diagnosis but are not exclusive for the disease, since they have already been identified in other inflammatory conditions of the lymph nodes. The immunohistochemistry reveals positive results for antigens CD1a, S-100 and CD4, but only the first one is exclusive and thus diagnostic for LCH (21).
Treatment
The ideal therapy for the LCH has not yet been set up. The variety of clinical manifestation of the disease and the fact that 10 to 20% of the patients have a spontaneous regression of the disease makes the comparative studies between different therapies difficult. Some authors suggest the treatment must be conservative and limited to the patients with constitutional symptoms such as fever, pain or dysfunction of target organs (lungs, liver, spleen and hematopoietic system) or otherwise based on the prognostic factors such as age, extension of the disease, affected regions and complications. The objectives of the treatment include improvement of the clinical symptoms and prevention of complications (21).
For unifocal disease like skin or bones lesions, the procedure is of expectation or the topical treatment is carried out. Especially in children younger than 1 year, the cutaneous lesions may regress spontaneously. When treatment is required, topical corticosteroids and in some cases nitrogenous mustard at 0.02% may be used. The local radiotherapy in low dosages is generally effective, but for the risk of malignancy induction its use is avoided in children (23). In the cutaneous lesions resistant to these modalities, a good choice is the systemic corticoid therapy in low dosages (24).
In the multifocal disease, the systemic medications are the main therapeutic option, separately or jointly. Amongst which, we include the systemic corticosteroids, cytostatic, at 2-chlorodesoxiadenosin (2CdA - cladribine) (25), cyclosporine (26) and in more recent studies, thalidomide (27) and myeloablative therapies. The indexes of response, recurrence and complications vary according to different schemes, but in general we verify the patients with target organs lesions have worse outcomes and a higher incidence of complications and death. In recurrent cases, the severity of the disease generally determines the therapy with a higher response probability (21).
FINAL COMMENTS
The Langerhans cell histiocytosis is an uncommon disease of children and its early manifestations occur mainly in the head and neck; its otorhinolaryngological manifestations must be suspected by recurrent otological symptoms and the presence of retroauricular or external auditory meatus granulation tissue. The biopsy with characteristic histological discoveries and immunohisto-chemistry positive for CD1a were diagnostic. Chemotherapy is the main form of treatment.
BIBLIOGRAPHICAL REFERENCES
1. Egeler RM. Historical Review - The Langerhans Cell Histiocytosis X Files Revealed. British Journal of Haematology. 2002, 116:3-9.
2. DiNardo LJ, Wetmore RF. Head and Neck Manifestations of Histiocytosis-X in Children. Laryngoscope. 1989, 99:721-724.
3. Willman CL, Busque L, Griffith BB, Favara BE, McClain KL, Duncan MH, et al. Langerhans-cell histiocytosis (histiocytosis X) - a clonal proliferative disease. N Engl J Med. 1994, 331(3):154-60.
4. Egeler RM, Favara BE, van Meurs M, Laman JD, Claassen E. Differential in situ cytokine profiles of Langerhans-like cells and T Cells in Langerhans Cell Histiocytosis: Abundant Expression of Cytokines Relevant to Disease and Treatment. Blood. 1999, 94(12):4195-4201.
5. Minkov M, Prosch H, Steiner M, Grois N, Pötschger U, Kaatsch P, et al. Langerhans cell histiocytosis in neonates. Pediatr Blood Cancer. 2005, 45(6):802-807.
6. Fuertes Cabero S, Fuster Pelfort D, Setoain Perego X, González Berenguer J, Mateos Fernandes JJ, Paredes Barranco P, et al. Usefullnes of bone scintigraphy for staging in a case of histiocytosis of the temporal bone. Rev Esp Med Nucl. 2005, 24(1):45-7.
7. Cajade Frías JM, Cajade Bao D, Lozano Ramírez A, Castro Vilas C, Vaamonde Lago P, Labella Caballero T. Unifocal eosinophilic granuloma of the temporal bone (Langerhans cell histiocytosis). Acta Otorrinolaringol Esp. 2000, 51(6):525-9.
8. Classificação Estatística Internacional de Doenças e Problemas Relacionados à Saúde. Décima Revisão. Organização Mundial de Saúde. 1997, Volume 1, 219;260-261.
9. Favara BE, Feller AC, Pauli M, Jaffe ES, Weiss LM, Arico M, et al. Contemporary classification of histiocytic disorders. The WHO Committee On Histiocytic/Reticulum Cell Proliferations. Reclassification Working Group of the Histiocyte Society. Med Pediatr Oncol. 1997, 29:157-166.
10. Cochrane LA, Prince M, Clarke K. Langerhans' cell histiocytosis in the pediatric population: presentation and treatment of head and neck manifestations. J Otolaryngol. 2003, 32(1):33-7.
11. Irving RM, Broadbent V, Jones NS. Langerhans' Cell Histiocytosis in Childhood: Management of Head and Neck Manifestations. Laryngoscope. 1994, 104:64-70.
12. Fernández-Latorre F, Menor-Serrano F, Alonso-Charterina S, Arenas-Jiménez J. Langerhans' Cell Histiocytosis of the Temporal Bone in Pediatric Patients: Imaging and Follow-up. Am J Radiol. 2000, 174:217-221.
13. Hudson WR, Kenan PD. Otologic Manifestations of Histiocytosis X. Laryngoscope. 1969, 25:678-693.
14. Hashimoto K, Schachner LA, Huneiti A, Tanaka K. Pagetoid self-healing Langerhans cell histiocytosis in an infant. Pediatr Dermatol. 1999, 16(2):121-7.
15. Ha SY, Helms P, Fletcher M, Broadbent V, Pritchard J. Lung involvement in Langerhans cell histiocytosis: prevalence, clinical features, and outcome. Pediatrics. 1992, 89(3):466-9.
16. Brown CW, Jarvis JG, Letts M, Carpenter B. Treatment and outcome of vertebral Langerhans cell histiocytosis at the Childrens Hospital of Eastern Ontario. Can J Surg. 2005, 48(3):230-236.
17. McCaffrey TV, McDonald TJ. Histiocytosis X of the Ear and Temporal Bone: Review of 22 cases. Laryngoscope. 1979, 89:1735-1742.
18. Anonsen CK, Donaldson SS. Langerhans' Cell Histiocytosis of the Head and Neck. Laryngoscope. 1987, 97:537-542.
19. Rosenzweig KE, Arceci RJ, Tarbell NJ. Diabetes insipidus secondary to Langerhans cell histiocytosis: is radiation therapy indicated? Med Pediatr Oncol. 1997, 29(1):36-40.
20. Risdall RJ, Dehner LP, Duray P, Kobrinsky N, Robison L, Nesbit ME. Histiocytosis X (Langerhans' cell histiocytosis). Prognostic role of histopathology. Archives of Pathology and Laboratory Medicine. 1983, 107:59-63.
21. Tebbi CK, Arceci RJ, Loew TW. Histiocytosis. Emedicine, 2002. Disponível em: http://www.emedicine.com/ped/topic1997.htm
22. Muscolo DL, Slullitel G, Ranalletta M, Aponte-Tinao LA, Ayerza MA. Spontaneous remission of massive solitary eosinophilic granuloma of the femur. J Pediatr Orthop. 2003, 23(6):763-765.
23. Kleijung T, Woenckhaus M, Bachthaler M, Wolff JEA, Wolf SR. Langerhans' Cell Histiocytosis with Bilateral Temporal Bone Involvement. Am J Otolaryngol. 2003, 24(4):265-270.
24. Yamaguchi S, Oki S, Kurisu K. Spontaneous regression of Langerhans cell histiocytosis: a case report. Surg Neurol. 2004, 62(2):136-140.
25. Saven A, Burian C. Cladribine Activity in Adult Langerhans-cell Histiocytosis. Blood. 1999, 93(12):4125-4130.
26. Mahmoud HH, Wang WC, Murphy SB. Cyclosporine therapy for advanced Langerhans cell histiocytosis. Blood. 1991, 77(4):721-725.
27. Wu JJ, Huang DB, Pang KR, Hsu S, Tyring SK. Thalidomide: dermatological indications, mechanisms of action and side-effects. Br J Dermatol. 2005, 153(2):254-273.
1. Doctor. Otorhinolaryngologist.
2. Master's Degree at the University of Brasília. Assistant Doctor, Preceptor and Coordinator of the Medical Residence Program in Otorhinolaryngology of the Hospital de Base do Distrito Federal.
3. Master's Degree at the University of Brasília. Assistant Doctor, Preceptor and Coordinator of the Medical Residence Program in Otorhinolaryngology of the Hospital de Base do Distrito Federal.
Institution: Hospital de Base do Distrito Federal. Brasília / DF - Brazil. Mail Address: Humberto de Barros Fernandes - Rua 24 de Janeiro, 2139 - Bloco H - Apto. 301 - Teresina / PI - Brazil - Zip Code: 64016-903 - Telephone: (+55 86) 9929-1019 / 9947-9844/ 3218-6173 - E-mail: humbertodbf@yahoo.com.br. Article received on July 22 2008. Approved on August 2 2009.
|
|
 |
|
|
