INTRODUCTIONMucopolysaccharidosis (MPS) is a group of rare diseases caused by the deficit of lysosomal enzymes, leading to an accumulation of glycosaminoglans (GAG) in organs and tissues, responsible for the multisystemic, chronic and progressive clinical features. 11 enzymatic deficits have been identified and cause 7 different types of MPS. The mucopolysaccharidosis treatment was initially symptomatic and palliative based on multidisciplinary team including a range of medical fields, such as cardiology, pneumology, anesthesia, orthopedics, otorhinolaryngology, ophthalmology, neurosurgery, among others, as well as professionals in physiotherapy, occupational therapy, psychology, and phonoaudiology (1) . There is presently a specific therapy for types I, II and VI in addition to the enzymatic replacement therapy (ART), consisting in a constant venous administration of the specific deficient enzyme on the patient. This treatment has achieved good results in both clinical improvement and quality of life (2).
MPS I, like most lysosomal diseases, is inherited in an autosomal recessive way and it has a 1:100,000 live newborns ratio (3). MPS I-related phenotypes can be shown such as Hurler, Hurler-Scheie and Scheie's Syndrome. Hurler's Syndrome is the most severe type of the disease and Scheie's Syndrome is the weakest one.
MPS II (Hunter's Syndrome) shows a ratio of nearly 0.31-0.71 per 100,000 live newborns (4, 5, 6), and it is exclusively found in boys since it is an X chromosome-associated genetic disease.
Mucopolysaccharidosis VI (MPS VI or Maroteaux-Lamy's Syndrome) is an infrequent autosomal recessive genetic disease caused by the deficit of the enzyme N-acetylgalactosamine-4-sulfatase, or arylsulfatase B. The estimated incidence for MPS VI is 0.23:100,000 live newborns.
MPS can be diagnosed according to clinical suspicion, measurement of positive urinary GAG by an enzymatic evaluation. In most cases, the disease appears during childhood and no clinical symptom is evident at birth. MPS patients show a wide range of multisystemic symptoms with a chronic and progressive course, impairing mostly the skeletal and cardiopulmonary systems, cornea, skin, liver, tobacco, brain and meninges.
The most common clinical otorhinolaryngological manifestations are respiratory anomalies and repetitive infections of the upper airways (UA) as a result of macroglossia, adenoids and amygdales hypertrophy, skeletal alterations of mandible and cervical column, sleep apnea, repetitive otitis and hearing loss. The otorhinolaryngological evaluation can precede the syndrome diagnosis in nearly 30% of the cases (7).
The group of otorhinolaryngological manifestations must be properly surveyed, in order to help reach both an early diagnosis of the syndrome and the specific treatment leading to a decrease in the co-morbidities, thus enhancing the quality of life. However, studies in literature about this group of patients are rare.
It is a transverse section study intended to describe the profile of the clinical otorhinolaryngological exam of patients submitted to ERT.
METHODAn otorhinolaryngological evaluation was performed in 21 patients with MPS using ERT.
Among the evaluated patients, 12 had MPS type VI, 6 type II and 3 type I. As to the patients with MPS type I, 1 resembles Hurler, 1 Hurler Scheie and 1 Scheie. Among MPS I and VI individuals, 12 were male. All MPS II patients were male. Age ranged between 1 and 20, and the average age was 9.14 years.
All the patients who were followed up with at the studied hospital and used ERT were included. Therapy length ranged from 7 to 60 months, at an average of 25.71 months.
Those responsible for the patients were interviewed and a physical evaluation of the patients was performed by otorhinolaryngologist doctors.
This study has been approved by the Ethical Committee under Resolution number 010/2009.
RESULTSAround 66% of patients showed a complaint of nasal obstruction, which was continual in 38% of patients and regarded as pronounced in 30% of cases. Snore was reported in 71% of patients, out of whom 50% had the symptom on a daily basis and a breathing pause was reported in 31% of cases. Snores were more frequent in MPS I and II patients, and MPS VI patients having snore complaints showed a higher intensity and a more frequent association with sleep apnea. Breathing was prevalently made by the mouth in 76% of patients. Half patients showed a rhinorrhea complaint, mostly in an intermittent (77.8%) and yellowed (55%) way.
The main type of UA infection was acute otitis media. Hearing loss was a symptom in only 9 cases (45%). Regarding MPS types, the frequency of symptoms can be compared in the Table 1.
As for the physical exam, TM retraction was the most frequent alteration in otoscopy (33%), out of which 30% showed a regular otoscopy at the moment of evaluation (Table 2). At the previous rhinoscopy, most patients showed no alteration.
At oropharyngoscopy, amygdales were not viewed in 60% of cases, an in cases with a good view, most of them were eutrophic. The grade of amygdala hypertrophy and the possibility of viewing oropharynx ranged with MPS types (Graphic 1).
Macroglossia was also verified in almost 100% of cases, pronounced in 40%. MPS II and VI patients showed a more pronounced macroglossia and all type I patients showed only slight macroglossia (Graphic 2).
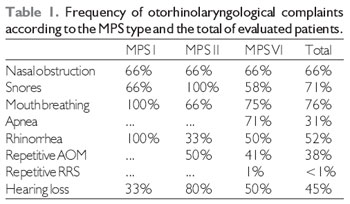
Legend: AOM - Acute otitis media RSA - repetitive rhinosinusitis.
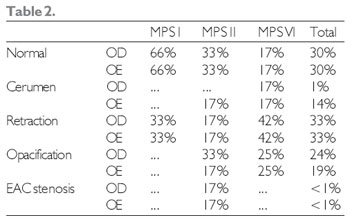
Legend: RE - right ear LE - left ear EAC - external auditory canal.
UA impairment is one of the most challenging aspects while treating these patients. UA alterations include macroglossia, adenoid amygdala hypertrophy, GAG deposit in pharynx and larynx. In an evaluation with 27 MPS patients, around 70% of them showed a clinical obstruction of the upper airways (8), according to the evaluation performed in our patients. The craniofacial alterations associated with the head and neck instability make them difficult to intubate, having a high anesthetic risk. At a 89-anesthetic procedure evaluation, difficulty in intubation was found in 25% of cases. Patients with Hurler's Syndrome showed twice as many difficulties. The decision about managing adenoid amygdala hypertrophy must be made based on the clinical features and polysomnography, due to the anesthetic risks of these patients.
The prevalence of audiologic alterations in MPS patients is still unknown. These patients show a highly frequent neurological impairment, and it is hard to reach a satisfactory audiometric evaluation. It is known that hearing loss is a frequent manifestation in these patients; in this study, it was reported in 40% of patients. This number can be even higher after audiometry is performed.
WOLD et al, while evaluating MPS I, II and VI patients, showed 78% of hearing loss by an audiometric evaluation. Out of these, 71% showed a mixed hearing loss, 14% sensorineural and 14% conductive (7). The histological evaluation of the temporal bone suggests that there is interference in the development around 5-6 months of pregnancy; there is subsequently a reduction in the mastoid pneumatization, persistence of cartilaginous capsule of the eye neighboring the posterior semicircular canal and an increase in the subarcuate artery (10). The conductive hearing losses are related to the presence of effusion and ossicular impairment in the middle ear (11). In this evaluation, the presence of effusion in these patients' middle ears has not been clearly verified, what can be explained by the age of evaluation, use of enzymatic replacement therapy or the opacification of the tympanic membrane. A long-term follow up and a comparison with patients not using this therapy are therefore required.
The etiology of the sensorineural hearing loss remains obscure and the possibility of nervous compression by arachnoids hyperplasia and axon destruction in the spiral ganglion is suggested.
Previous studies (11) show a hearing loss more frequently in MPS I and II patients, such data is different from those indicated in this evaluation. This difference can have happened as a result of the higher number of MPS VI patients included in this study.
In a retrospective evaluation with 09 MPS I, II, III patients aged around 3, it was verified that all of them needed to be submitted to a VT placement, due to the otitis media with effusion/repetitive acute otitis media. Half of these patients was submitted to an initially conservative treatment, without a solution; as for the patients who chose a short-term venting tube, the process had to be repeated; nonetheless, the author recommends that the long-term venting tube to be chosen.
In our service, the following flowchart drawn in Figure 1 is being performed from the first evaluation to the diagnosis proposal and subsequent intervention. The data mentioned in this article represents the first otorhinolaryngological evaluation of these patients submitted to more detailed hearing and upper airway evaluations.
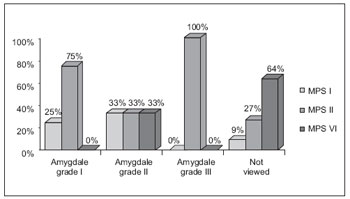
Graphic 1. Comparison of the oropharynx evaluation in relation to MPS I, II, VI.
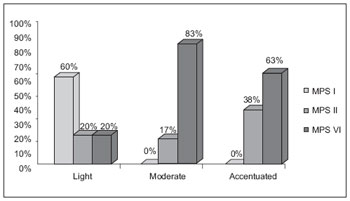
Graphic 2. Comparison of the macroglossia degree in MPS I, II, VI.
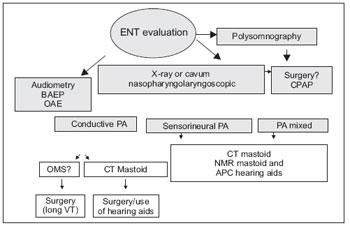
Figure 1. BAEP - Brainstem Auditory Evoked Potential OAE - Otoacoustic Emissions CT - Computed Tomography NMR - Magnetic Resonance VT - venting tube CPAP - Continuous positive airway pressure.
Following up with these patients requires a multidisciplinary team to cooperate, and the documents about the pathology evolution must be provided based on follow-up and diagnostic records. The otorhinolaryngological evaluation and follow-up must be at an early stage, in order to prevent major systemic impairments in the changes of the upper airways and irreversible damages in hearing.
REFERENCES1. Pastores GM, Arn P, Beck M, Clarke JT, Guffon N, Kaplan P, et al. The MPS I registry: design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Mol Genet Metab. 2007, 91(1):37-47.
2. Giugliani R, Federhen A, Munoz Rojas MV, Vieira TA, Artigalas O, Pinto LL, et al. [Enzyme replacement therapy for mucopolysaccharidoses I, II and VI: recommendations from a group of Brazilian F experts.]. Rev Assoc Med Bras. 2010, 56(3):271-7.
3. Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999 Jan 20, 281(3):249-54.
4. Baehner F, Schmiedeskamp C, Krummenauer F, Miebach E, Bajbouj M, Whybra C, et al. Cumulative incidence rates of the mucopolysaccharidoses in Germany. J Inherit Metab Dis. 2005, 28(6):1011-7.
5. Nelson J, Crowhurst J, Carey B, Greed L. Incidence of the mucopolysaccharidoses in Western Australia. Am J Med Genet A. 2003 Dec 15, 123A(3):310-3.
6. Nelson J. Incidence of the mucopolysaccharidoses in Northern Ireland. Hum Genet. 1997, 101(3):355-8.
7. Wold SM, Derkay CS, Darrow DH, Proud V. Role of the pediatric otolaryngologist in diagnosis and management of children with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2010, 74(1):27-31.
8. Andrea H.Yeung M, Morton J.Cowan M, Biljana Horn M, Kristina W. Rosbe M. Airway Management in Children With Mucopolysaccharidoses. Arch Otolaryngol Head Neck Surg. 2009, 135(1):73-79.
9. Walker RW DMMPWJE. Anaesthesia and mucopolysaccharidoses. A review of airway problems in children. Anaesthesia. 2010, 49(12):1078-1084.
10. Hayes E, Babin R, Platz C. The otologic manifestations of mucopolysaccharidoses. Am J Otol. 1980, 2(2):65-9.
11. Papsin BC, Vellodi A, Bailey CM, Ratcliffe PC, Leighton SE. Otologic and laryngologic manifestations of mucopolysaccharidoses after bone marrow transplantation. Otolaryngol Head Neck Surg. 1998, 118(1):30-6.
12. Motamed M, Thorne S, Narula A. Treatment of otitis media with effusion in children with mucopolysaccharidoses. Int J Pediatr Otorhinolaryngol. 2000, 53(2):121-4.
1) Specialist in Otorhinolaryngology. Fellow in Otology.
2) Specialist in Otorhinolaryngology. Deputy Professor of Otorhinolaryngology of Medical School of Bahia.
3) Graduated in Phonoaudiology. Taking Master degree in Biotechnology in Health and Investigative Medicine at Oswaldo Cruz School - Fiocruz.
4) Doctor in Medical Sciences. Professor of Pediatrics atthe Medical School of Bahia. Head of the Medical Genetics Department at the Professor Edgard Santos University Hospital Complex.
Institution: Professor Edgard Santos University Hospital Complex, Salvador / BA - Brazil. Mailing address: Cibele Gomes Bicalho - Rua Augusto Viana S/N - Canela - Salvador / BA - Brazil - ZIP Code: 40110-060 - Email: cibelebicalho@gmail.com
Article received on January 3, 2011. Article approved on April 7, 2011.

