INTRODUCTIONTreacher Collins Syndrome (TCS) or Mandibulofacial Dysostosis is a hereditary disorder which is characterized by craniofacial anomalies and presents several clinical variable manifestations.(1)
Firstly described by Thomson in 1846, it had in the Treacher Collins's approach the description of its essential parts, thus making its eponymic name the most used one in the English Literature.(1)
Its incidence is around 1:40,000 to 1:70,000 cases per live-born children, and there is no difference between genders or among races. Its transmission occurs by accident, but a dominant autosomal transmission with variable expressivity is suspected(2,3). A child has 50% of chances of inheriting such condition when one of its progenitors presents such syndrome(4).
The gene which carries the genetic alteration has been mapped in the distal portion of the chromosome 5 long arm (5q31.3-q33.3).(5) The phenotypical expression of such disease is probably the result of a congenital malformation which bilaterally involves the first and the second branchial archs(6,7). In 1949, Franceschetti and Klein suggested a basic pathogeny of such disease and said that such deficiency occurs during the seventh week of gestation, when the facial bones are under formation and the genetic inhibitory action may happen(8,9).
Such hereditary disorder contains antimogoloid slant of the palpebral fissures, coloboma of inferior palpebra, micrognatia and hypoplasia of the zigomatic and microtia archs. The symptoms which may be present in the clinical picture are: descendent slant of the palpebral fissures, palpebral ptosis, coloboma of inferior palpebras, hypoplasia of the malar and zygomatic eminences, maxillary hypoplasia with fissure of secondary palate or arched and high palate, absence of velum (rarely with choanal atresia) and extension of "temporal hair tongue" along the cheek sides. The labial fissures are rare(1,10,11).
What has motivated us to a deeper study of the Treacher Collins Syndrome is the interdisciplinary participation which is present in the daily routine of Hospital Universitário Bettina Ferro de Souza - Universidade Federal do Pará and Universidade do Estado do Pará covering the specialties of Otorrhinolaryngology, Ophtalmology, Neurology, Pediatrics, Dentistry, Psychology, Social Service and Physical Therapy, mainly concerning the assistance of patients who carry special needs. The achievement of this work is justified by the need to warn health professionals to the occurrence of such syndrome, which is little frequent but has serious consequences to its carriers, mainly when they are exposed to a late diagnosis.
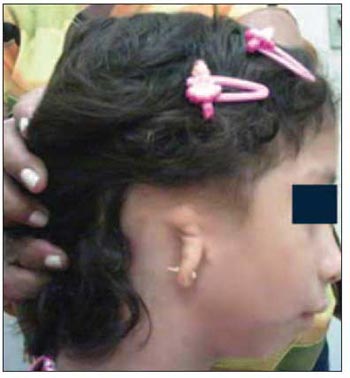
Picture 1. Treacher Collins Syndrome: malformation of auricular pavilion. - Reproduced from: Cavalcante HA, Moura WJQ, Oliveira LS, Lima BP, Mazivieiro SNA, Nunes CTA. Treacher Collins Syndrome associated to Chiari Malformation Kind I. IX North-Northeast Congress of Otorrhinolaryngology and Cervical-Facial Surgery, September 6 to 8, 2007, Belém, Pará. Poster.
To carry out a literature review about the several aspects of the Treacher Collins Syndrome, thus emphasizing the otorrhinolaryngological clinical manifestations.
MATERIAL AND METHODThe investigation through on line data provided information and articles to carry out this bibliographic review. Such articles may be accessed at any time to research and updating, once they are renewed as the scientific literature is produced.
The following data bases have been referred: Cochrane, LILACS, MEDLINE, OMIM and SciELO. The terms Treacher Collins Syndrome and Mandibulofacial Dysostosis have been applied to the investigation for articles published between the years of 1997 and 2007, in addition to famous literature related to such topic.
LITERATURE REVIEW
General Aspects and considerations concerning diagnosisThe gene which causes TCS has been mapped in the chromosome 5 long arm (5q31-33), being named TCOF1(12,13) made of 26 exons, out of which 25 are translated into a low-complexity protein, called treacle(14). The function of such protein is still unknown; however, it is suspected that it acts on the transportation of other proteins from the cytoplasm to the nucleus. It is believed that such individuals which suffer from TCS produce a protein which is shorter and more unstable than normal, in a way which the clinical picture may be caused by haploinsufficiency, that is, insufficient quantity of such protein(14,15). The exons which will most probably present deletions and insertions are the 10, 15, 18, 23 and 25 ones.(16) Consequently, such exons must be the first ones to be tested in any molecular study in patients which carry TCS. The mutations are caused by alterations in the sequence of the gene which affects the protein which is originated by such gene, and the deletion of the exon 24 of the gene TCOF1 is the most frequent one. The identification of the place and of the kind of mutation is important for the genetic advice of siblings and parents of the carrier patient(1,9,17).
The clinical expression of the disease is very variable, which ranges from mild manifestations up to severe cases, which may lead to misleading syndromic diagnosis. However, if two children of the same parents are considered, the diagnosis becomes more evident. Patients who present classical characteristics of TCS are may have their diagnosis relatively easily carried out(18).
These children must be analyzed at birth by an otorrhinolaryngologist and by a speech therapist, so that the extension of hearing loss and the degree or airways endanger are identified. In the individuals who are mildly affected, the diagnosis needs genetic evidence(19). Such diagnosis may be done during the antenatal care through a biopsy of the chorionic villus between the 10th and the 13th week or a amniocentesis between the 16th and the 18th week of gestation to study the genetic material. The ecography and the fetoscopy are auxiliary methods of such diagnostic process(19,20).
Some authors consider TCS a neonatal emergency in a way which, once it is detected, care must be taken in order to avoid the glossoptosis and the consequent obstruction of the superior air ways during the antenatal period, and ventilation and intubation may be necessary in the delivery room(21,22). The accurate diagnosis after the birth may be performed with DNA analysis of the syndrome carrier. The DNA analysis of the progenitors is also advisable for purposes of genetic advice(15,23).
Otorrhinolaryngological Clinical Manifestations and others Most of head and neck congenital malformations occur during the phase of branchial apparatus transformation and are located in certain portions of such apparatus, are not frequent and disappear as the definitive structures develop(24).
The remnant malformations originate the syndromes with mandibulofacial dysostosis, among which Treacher Collins Syndrome is the most common one. The most frequent TCS manifestations are closely related to the degree of facial deformity in the affected individuals, in whom the modifications which may be detected, followed or modified by otorrhinolaryngologists are many, as follows: antimongoloid slant of palpabral fissures (89%); hypoplasia of malar region, with or without fissure of the zygomatic bone (81%); mandible hypoplasia (78%) with variable effects to ATM and mastication muscles; coloboma of inferior palpebras (69%); partial or complete absence of eyelash in the inferior palpebras (53%); malformation of auricular pavilions (36%); atresia of the external auditory canal (40%); conductive deafness (28%); presence of hair in the external part of face (26%) and palatine fissure (32%)(1,25).
Some deformities are less frequently related to such picture, such as the pharynx hypoplasia; narrow nostrils with nasal dorsus with parrot-like shape; high palate; microphtalmia; macrostomia or microstomia; choanal atresia; bottom-bag fistulas and Skin appendix between the auricular pavilion and mouth angle(1,26,27).
Malformations with repercussion in other parts of the body may also occur like the congenital cardiopathy, the cryptorchidism and mental deficiency(1).
The malformations which are diagnosed at birth are stable, that is, are not progressive according to age.(19) The obstruction of superior air ways due to structural factores mat frequently cause obstructive apnea in TCS carriers, like in the case of the base angle on reduced skull placing the pharynx from back to front; of narrow nasopharynx and of maxilla - vertically reduced with reduced anterior projection - reducing the antero-posterior dimension of nasopharynx even more.(1) The present micrognathia related to tongue in posterior position generates modifications which cause an improper respiratory standard(28).
The craniofacial skeleton presents abnormalities in the mandible, maxilla, zygomatic areas, orbits, ears and skull base. The height of the mandibular branch is deficient and the length of the mandible body is reduced. The distortion which is present in the symphysis menti contributes for the mandibular deficiency and for the increase of the inferior height of the face, which leads to an increase of the cranial-base-mandibular angle. The mentum angle may also be bigger than the standard. Consequently, the mandible in TCS carriers is retrognatic, the temporal-mandibular articulation is anteriorly misplaced, the mandibular angle is obtuse and the mandible is smaller than the maxilla(26).
Differential DiagnosisThe differential diagnosis is important in every craniofacial deformity, and in such case it should include the following syndromes:
a) 1st branchial arch syndrome (Oral-mandibular-auricular syndrome). Macrosthomia, hemignathia and tragus abnormalities appear in this syndrome, as well as in the superior part of the helix, once both portions are originated from the first arch(9,29,30).
b) 1st and 2nd arch syndrome. Hemignatia and deformities I all the auricular pavilion are observed. The 1st and the 2nd branchial arcs are joined in the superior half of the helix, in the mandible and in the temporal-mandibular articulation(9,29,30).
c) Other anomalies, such as the Crouzon Disease, Apert Syndrome, hyperterolism, Möbius Syndrome and Romberg disease should always make the differential picture(9,29).
Evolution and TreatmentIts treatment starts in the antenatal phase, through family advice in terms of the particularities of such anomaly and necessary care during life.
These patients may present respiratory difficulties in the first months of life caused by the presented malformations, which may become complicate with colds and infections, mainly in children. The use of temporary tracheotomy and CPAP is very favorable. Intubation is usually difficult due to narrow air ways(11,22,31).
If the patient carries palatine fissure, it should be dealt according to the current treatment protocols, with the immediate use of functional orthopaedics of the maxillas extending as much as possible the surgical procedure of fissure closing until the beginning of alphabetization(1,4,11,32).
Even if there is no fissure, if the maxilla orthopaedics is done through fixed orthodontics, it greatly prevents or reduces the dental-facial deformities features of Manidbulofacial Dysostosis(1,11).
The mandibular problem must be faced as if the patient carried craniofacial microsomia, thus following the protocol for mandibular distraction, and the eruption of the first permanent molar teeth are one of the great indications of bilateral mandibular distraction(1,11,32).
The early diagnosis of hypoacusis or deafness and its early correction are essential, once the delay of intellectual development is not such a frequent characteristic. Surgery may be used for that aim, or deafness correction with auditory devices, which contributes for the individual proper development(25,30).
The growth of facial bones during the first and second infancies causes certain improvement in the aesthetic conditions, which may improve even more with plastic surgery(3).
The treatment of facial hard and soft tissues may require a great number of surgeries: first, the correction of palpebra coloboma in the first years of life(33); after that, the orbit reconstruction with calvaria bone grafts and correction of lateral canthal displacement; reconstruction of ear is done from 5 to 7 years of age; then, the correction of the inferior part of face and mandible, by craniofacial surgeon, pediatric dentist and orthodontist, with the orthodontic intervention in the beginning of the eruption of the patient's permanent teeth. After teeth are aligned in their axis, the treatment of the inferior face repositions the mandible and the maxilla, at around 10 years of age. As the child's face continues growing, additional procedures may be required to correct and development deformity(24,32).
FINAL CONSIDERATIONSTreacher Collins Syndrome must continue being investigated, so that it is possible to improve the current approach in functional and aesthetical corrections, improving the psychosocial support and the participation of multidisciplinary team made of otorrhinolaryngologists, craniofacial surgeons, ophthalmologists, speech therapists, psychologists and oral surgeons. Findings of genetic alterations have constantly being done, greatly enabling the early diagnosis and the familiar genetic advice, contributing for the improvement of the treatment and of the diagnosis, reducing the social stigma and offering life quality to patients and their families.
REFERENCES1. Bezerra SMP, Ortega AOL, Guaré RO, et al. Síndrome de Treacher Collins: características clínicas e relato de caso. Rev Pos Grad. 2005, 12(4):499-505.
2. Hungria, H. Disacusias. Implante coclear. A criança surda. In: Otorrinolaringologia. 8 ed. Rio de Janeiro: Guanabara Koogan; 2000.
3. Nora JJ, Fraser FC. Doenças mendelianas selecionadas. In: Genética Médica. 3 ed. Rio de Janeiro: Guanabara Koogan; 1991.
4. Posnick JC, Ruiz RL. Treacher Collins syndrome: current evaluation, treatment and future directions. Cleft Palate Craniofac J. 2000, 37(5):434-64.
5. Arn PH, Mankinen C, Jabs EW. Mild mandibulofacial dysostosis in a child with a deletion of 3p. Am J Med Genet. 1993, 46:534-536.
6. Argenta LC, Iacobucci JJ. Treacher Collins syndrome: present concepts of the disorder and their surgical correction. World J Surg. 1989, 13(4):401-9.
7. Shah FA, Ramakrishna S, Ingle V, et al. Treacher Collins syndrome with acute airway obstruction. Int J Pediatr Otolaryngol. 2000, 54(1):41-3.
8. Andrade CE, Júnior VS, Didoni ALS, et al. Síndrome de Treacher Collins com atresia coanal: relato de caso e revisão de suas características. Rev Bras Otorrinolaringol. 2005, 71(1):107-10.
9. Carpes LCF, Avelino LC. Disostose Mandíbulo - Facial. Rev Bras Otorrinolaringol. 1974, 40(1):57-60.
10. Dixon MJ. Treacher Collins syndrome. Hum Molec Genet. 1996, 1391-1396.
11. Bergonse GFR, Carneiro AF, Vassoler TMF. Atresia de coana: análise de 16 casos - a experiência do HRAC-USP de 2000 a 2004. Rev Bras Otorrinolaringol. 2005, 71(6):730-3.
12. Su PH, Yu JS, Chen JY, et al. Mutations and new polymorphic changes in the TCOF 1 gene of patients with oculo-auriculo-vertebral spectrum and Treacher-Collins síndrome. Cin Dysmorphol. 2007, 16(4):261-7.
13. Dixon MJ. Treacher Collins syndrome. J Med Genet. 1995, 32(10):806-8.
14. Treacher Collins Syndrome Collaborative Group. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nature Gene. 1996, 12:130-136.
15. Bueno MRP, Splendore A. Síndrome de Treacher Collins: Aspectos clínicos, genéticos e moleculares. Rev Med São Paulo. 2001, 80(1):52-56.
16. Aguiar RS, Santos CS. Síndrome de Treacher Collins. Rev Port Estomatol Cir Maxilofac. 1989, 30(2):137-42.
17. Wise CA, Chiang LC, Paznekas WA, et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins syndrome throughout its coding region. Proc Nat Acad Sci. 1997, 94:3110-3115.
18. Dixon MJ. Treacher Collins Syndrome: from linkage to prenatal testing. J Laryngol Otol. 1998, 112(8):705-709.
19. Posnick JC. Treacher Collins Syndrome: perspectives in evaluation and treatment. J Oral Maxillofac Surg. 1997, 55(10):1120-1133.
20. Ruangvutilert P, Sutantawibul A, Susaneevithayaku IP, et al. Ultrasonographic prenatal diagnosis of Treacher Collins syndrome: a case report. J Med Assoc Thai. 2003, 86(5):482-8.
21. Ochi H. Prenatal sonographyc diagnosis of Treacher Collins syndrome. Obstet Gynecol. 1998, 91(5):862.
22. Jayasekera N. Laryngeal mask for airway management in Treacher-Collins syndrome. Ceylon Med J. 2007, 52(1):34.
23. Ellis PE, Dawson M, Dixon MJ. Mutating testing in Treacher Collins Syndrome. J Orthod. 2002, 29(4):293-98.
24. Moore KL. Aparelho branquial, cabeça e pescoço. In: Fundamentos da embriologia humana. São Paulo: Manole.25. Smith DW. Síndrome de Malformações Congênitas. 3ª ed. São Paulo: Manole; 1985.
26. Bhatia S, et al. Radiocephalometric evaluation of a family with mandibulofacial dysostosis. Am J Orthod Dentofacial Orthop. 1996, 110(6):618-623.
27. Wiedemann H. Atlas de síndromes Dismórficas. 3ª ed. São Paulo: Manole; 1992.
28. Arvystas M, Shprintzen RJ. Craniofacial morphology in Treacher Collins syndrome. Cleft Palate Craniofac J. 1991, 28(2):226-301.
29. Roy FH. Diagnóstico diferencial em oftalmologia. 2ª ed. São Paulo: Manole; 1978.
30. Granström G, Jacobsson C. First and second branchial arch syndrome: aspects on the embryogenesis, elucidations, and rehabilitation using the osseointegration concept. Clin Implant Dent Relat Res. 1999, 1(2):59-69.
31. Negamine Y, Kurahashi K. The use of three-dimensional computed tomography images for anticipated difficult intubation airway evaluation of a patient with Treacher Collins syndrome. Anesth Analg. 2007, 1005(3):626-8.
32. Mccarthy JG, Hopper RA, Hollier LH. Molding of the regenerate in mandibular distraction: clinical experience.Plast Reconstr Surg. 2003, 112(5):1239-46.
33. Reiff ABM, Cunha KN, Figueiredo JCA, et al. Tratamento do coloboma de pálpebra superior pela técnica de Cutler- Beard e sua associação com ambliopia - Relato de caso. Arq Bras Oftalmol. 2002, 65(5):579-84.
34. Cavalcante HA, Moura WJQ, Oliveira LS, Lima BP, Mazivieiro SNA, Nunes CTA. Síndrome de Treacher Collins associada a Malformação de Chiari do Tipo I. IX Congresso Norte-Nordeste de Otorrinolaringologia e Cirurgia Cérvico-Facial, 06 a 08 de setembro de 2007, Belém, Pará. Pôster.
1. Otorrhinolaryngology Tutor at Universidade do Estado do Pará. Medicine senior student.
2. Assistant professor of Universidade do Estado do Pará. Scientific coordinator and preceptor of Otorrhinolaryngology residence at Hospital Universitário Bettina Ferro de Souza of Universidade Federal do Pará.
3. Ophtalmology tutor of Universidade do Estado do Pará. Medicine senior student.
4. Assistant professor of Universidade do Estado do Pará. Scientific coordinator of the Hearing Health Program at Hospital Universitário Bettina Ferro de Souza of Universidade Federal do Pará.
Universidade do Estado do Pará
Dorivaldo Lopes da Silva
Address: Trav. Dom Romualdo de Seixas ,1630, apto 404 - Belém/PA - CEP: 66055-200 - Phone: (91) 3225-0158 / (91) 9185-2065 - E-mail: dorivaldols@yahoo.com.br
This article was submitted at SGP (Publication Management System) of R@IO on December 23, 2007 and approved on February 1st, 2008, 18:36:45

