INTRODUCTIONRichner-Hanhart's syndrome or Tyrosinemia type II is characterized by the absence in a variable degree of distal portions of one or more extremities, in association with micrognathia and severe microglossia or both (1).
The etiology of this uncommon syndrome remains unknown and both environmental and genetic factors have been regarded (2). Estimated prevalence is 1:500.000 (1).
The pathogenesis of the tissue damage is not very clear. High plasmatic levels of tyrosine and metabolites (phenolic acids) have been related, but this association has not been confirmed yet (3).
As differentiated diagnoses for this syndrome, ectrodactyly, isolated cranial nerve palsy and Poland's Syndrome must be considered (1).
The dietary treatment for this pathology causes the correction of chemical abnormalities (tyrosinemias) and a significant enhancement of the cutaneous and ocular lesions. The renal and growth affection can as well be prevented or enhanced, but the progression of liver impairment cannot be refrained (1).
LITERATURE'S REVIEWRichner-Hanhart's Syndrome or Tyrosinemia type II (Oculocutaneous Tyrosinemia) is a rare autosomal recessive disorder associated with the tyrosine metabolism, characterized by the deficit of cytosol fraction of Hepatic Tyrosine Aminotransferase (TAT). The responsible gene is found in the chromosome 16q22.1-q 22.3. Under 100 cases were documented, the majority of which was observed in Italians (3).
The first case of congenital tongue agenesis was described by De Jussieu in 1718, on a 15-year-old girl. Kettner, in 1907, was the first to associate aglossia with the various hand and foot abnormalities on a 4-year-old child. Shortly after 70 years, in 1975, nearly 20 cases had been counted in the world's literature (1).
The association of punctiform palmoplantar keratodermas, keratitis and mental retardation was firstly and independently described by RICHNER (1938) and HANHART (1947), but the relation with tyrosine metabolism had not been discovered until 1973 (3).
The etiology of this uncommon syndrome remains unknown and environmental and genetic factors are studies (2).
In Richner-Hanhart's syndrome, the severity ranges with the significance of the different impaired extremities, from complete peromelia to the distal absence of a finger, syndactyly or ungual hypoplasis. Micrognathia and microglossia can be severe and associated with palatine fissure, synechias, syngnathia and oligodontia. Congenital palsy of unilateral or bilateral cranial nerves may occur. Intellectual ability is usually maintained (1).
There are 3 types of tyrosinemia and the clinical and biochemical deficiencies are different from each other (3).
In type I, there is a severe impairment of liver, kidneys and the central nervous system (3). It is associated with a growth deficit, rickets, Fanconi's Syndrome, progressive liver failure, higher levels of tyrosine and methionine in blood, as well as an increase in the urinary excretion of tyrosine and metabolites (tyrosinuria) and gamma-aminolevulinic acid. A deficit in the activities of the liver enzymes, such as TAT and 4-hydroxyphenylpyruvate dioxygenase, has been described. The deficit of fumarylacetoacetate hydrolase has been recently demonstrated in a number of patients and regarded as the primary enzymatic deficit found in this disease. NTBC and liver transplant are required for the treatment of this type.
Tyrosinemia type 2, described in nearly 20 patients, most of whom having Richner-Hanhart's Syndrome, associated with constant severe keratitis and hyperkeratosis mostly diverse in the palms of hands and the soles of the feet, shows a comprehensive response to the tyrosine-restricted diet. Blood tyrosine is found very high and there is a significant tyrosinuria. Absence of liver cytosol TAT has been demonstrated in 2 patients and an intermediate deficit was evident in 2 other patients, in the liver and fibroblasts, respectively (3).
Type 3 is extremely rare and the neurological association is prevailing (3).
CASE REPORTLLSS, male, 1 year and 11 months old, single, brown skin, born in the city of Itaperuna / RJ and residing in the city of São José de Ubá / RJ. When he was born, the pediatrician noticed that not only had the newly-born (NB) not shown an adequate suction reflex but he could only suck through a nursing bottle. Accordingly, he decided to submit the baby to an otorhinolaryngologist. At oroscopy, the NB was observed to have an agenesis of a back third of the tongue in association with micrognathia; malformation of feet, hands, fingers and toes (Figures 1, 2, 3 and 4); nasal septum deviation to the left and absence of adenoid. His larynx had no abnormalities.
X-rays of hands and feet were requested and showed a fusion of bones in these organs.
When analyzing all the clinical characteristics noticeable in the NB of this case report, it was concluded that he had a rare syndrome called Richner-Hanhart's Syndrome.
In an attempt to understanding the likely etiology of this syndrome, his mother was inquired about the possibility of consanguineous marriage and the use of illicit drugs during pregnancy, and both hypotheses were denied by her. The NB was, therefore, submitted to the geneticist to clarify the causes of his syndrome.
The infant is currently followed up with by the otorhinolaryngologist and submitted to a physiotherapy treatment. His speech was normal and his motor development met the normality standards for his age.
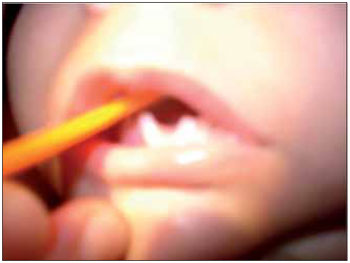
Figure 1. Image showing an agenesis of the back third of the tongue.
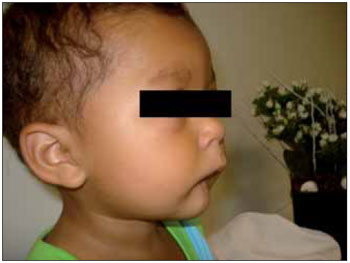
Figure 2. Image making micrognathia evident.
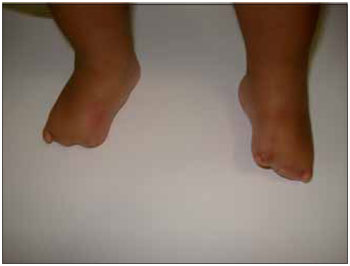
Figure 3. Image showing an abnormality of the toes.
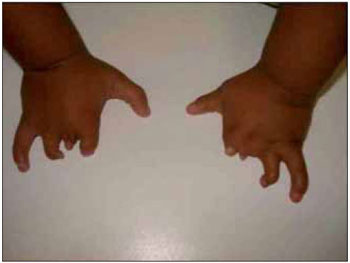
Figure 4. Image showing an abnormality of the fingers.
The etiology of this syndrome is unknown. Evidences of family heritage were not found and the hereditary factors would be irrelevant. Some authors defend that this pathology is likely to occur by autosomal recessive inheritance, consanguinity or autosomal dominant inheritance. However, others consider that environmental factors may cause this condition (1).
This condition is usually evident during infancy, with cutaneous lesions (85% of cases), ocular symptoms (75%) or neurologic complications (60%) or any association of the three (3).
The ocular manifestations usually start around the 2nd week of life, whereas the skin manifestations occur after the first year, but, in some cases, it can also occur before one month of life (3).
Due to the micrognathia, alterations in the sucking pattern can happen and they require an early intervention, because they can impair the chewing and deglutition functions (8). As another participation of the tongue besides chewing, speech can also be impaired (9).
A low tyrosine and phenylalanine diet in association with vitamin C reduce the serial levels of tyrosine. In some patients, however, refraining the disease from evolving is not possible by a diet only, hence, a liver transplant can be necessary (6).
CONCLUSIONRichner-Hanhart's Syndrome is extremely uncommon. It is, however, important to describe it so that it can be recognized and diagnosed beforehand, as well as further studies can be performed to clarify their likely etiologies, whether hereditary or environmental.
REFERENCES1. Castillo ST, Rojas JZ, Monasterio LA. Sindrome de Hanhart. Rev Chil Pediatr. 1985, 56(3):180-183.
2. Gillerot Y, Maldergem LV, Chef R, Koulischer L. Hypoglossia-hypodactyly syndrome with hydrocephalus: a clue to the aetiology? J Med Genet. 1991, 28: 490-491.
3. Paige DG, Clayton P, Bowron A, Harper JI. Richner-Hanhart syndrome (oculo cutaneoustyrosinaemia, tyrosinaemia type II). Journal of the Royal Society of Medicine. 1992, 85:759-760 .
4. Janakiraman L, Sathiyasekaran M, Deenadayalan M, Ganesh R, Mahesh U. Richner Hanhart Syndrome. Indian Journal of Pediatrics. 2006, 73:161-162.
5. Ramos BE, Pascual MMJ. Tratamiento dietético de las enfermedades metabólicas. Informacion terapeutica del sistema nacional de salud. 2005, 29(4):81-95.
6. Monroy LC, Urbanes MB, Sedan CA. Tirosinosis: Reporte de un caso. Revista Colombiana de Padiatría.
7. Cantani GO, Kennaway A, D'eufemia NGP. Tirosinemia crônica associada com deficiência de 41-hidroxifenilpiruvato dioxigenase com ataxia aguda intermitente e sem envolvimento visceral e ósseo. Pediatr. Res. 1983, 17:25-29.
8. Silva RCL, Alves FFS, Netto SSG, Silva CM. As alterações fonoaudiológicas na síndrome de Goldenhar - relato de caso. Rev Soc Bras Fonoaudiol. 2008, 13(3):290-5.
9. Freitas AC, Filho PN, Queiroz AM, Assed S, Silva FWGP. Síndrome de Moebius: Relato de caso clínico. Revista de Odontologia da Universidade Cidade de São Paulo. 2006, 18(3):297-302.
1) Specialized in Otorhinolaryngology at the Clinical Hospital in Itaperuna.
2) Internship student in Otorhinolaryngology. Undergraduate student in the sixth year of Medicine.
3) Junior Doctor in Otorhinolaryngology at São José do Avaí Hospital / RJ.
4) Undergraduate student in the sixth year of Medicine.
5) Internship student in General Surgery at São José do Avaí Hospital / RJ. Undergraduate student in the sixth year of Medicine.
Institution: Clinical Hospital. Itaperuna / RJ - Brazil. Mailing address: Daniela Silva Pais - Rua Rui Barbosa 383, 602 - Itaperuna / RJ - Brazil - ZIP Code: 28300-000 - Telephone: (+55 22) 8801-5160 - Email: danielasilvapais@gmail.com
Article received on August 24, 2009. Article approved on January 27, 2010.

